Arimidex 1mg Anastrozole Gebruik, bijwerkingen en dosering. Prijs in online apotheek. Generieke medicijnen zonder recept.
Wat is Arimidex en hoe wordt het gebruikt?
Arimidex 1 mg is een receptgeneesmiddel dat wordt gebruikt voor de behandeling van de symptomen van borstkanker bij postmenopauzale vrouwen. Arimidex kan alleen of in combinatie met andere medicijnen worden gebruikt.
Arimidex behoort tot een klasse geneesmiddelen die antineoplastische middelen, aromataseremmer worden genoemd.
Het is niet bekend of Arimidex veilig en effectief is bij kinderen.
Wat zijn de mogelijke bijwerkingen van Arimidex?
Arimidex kan ernstige bijwerkingen veroorzaken, waaronder:
- kortademigheid,
- bot fractuur,
- opgezwollen klieren,
- misselijkheid,
- pijn in de bovenbuik,
- jeuk,
- vermoeidheid,
- verlies van eetlust,
- donkere urine,
- kleikleurige ontlasting,
- geel worden van de huid of ogen (geelzucht),
- plotselinge gevoelloosheid of zwakte (vooral aan één kant van het lichaam),
- plotselinge ernstige hoofdpijn,
- onduidelijke spraak,
- problemen met zicht of evenwicht,
- koorts,
- keelpijn,
- zwelling in uw gezicht of tong,
- brandend in je ogen,
- huidpijn, en
- rode of paarse huiduitslag die zich uitbreidt (vooral in het gezicht of bovenlichaam) met blaarvorming en vervelling
Roep meteen medische hulp in als u een van de bovenstaande symptomen heeft.
De meest voorkomende bijwerkingen van Arimidex zijn:
- zwakheid,
- opvliegers,
- gevoelloosheid of tintelend gevoel in uw huid,
- zwelling in uw enkels of voeten,
- gewrichtspijn of stijfheid,
- problemen met uw vingers tijdens het grijpen,
- keelpijn,
- hoofdpijn,
- rugpijn,
- bot pijn,
- depressie,
- stemmingswisselingen,
- slaapproblemen (slapeloosheid),
- hoge bloeddruk,
- ernstige hoofdpijn,
- wazig zien,
- bonzen in je nek of oren,
- misselijkheid,
- braken, en
- milde uitslag
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van Arimidex. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
OMSCHRIJVING
ARIMIDEX (anastrozol) tabletten voor orale toediening bevatten 1 mg anastrozol, een niet-steroïde aromataseremmer. Het wordt chemisch beschreven als 1,3-benzeendiacetonitril, a, a, a', a'-tetramethyl-5-(1H-1,2,4-triazool-1-ylmethyl). De molecuulformule is C17H19N5 en de structuurformule is:
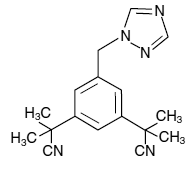
Anastrozol is een gebroken wit poeder met een molecuulgewicht van 293,4. Anastrozol heeft een matige oplosbaarheid in water (0,5 mg/ml bij 25°C); oplosbaarheid is onafhankelijk van de pH in het fysiologische bereik. Anastrozol is goed oplosbaar in methanol, aceton, ethanol en tetrahydrofuran en zeer goed oplosbaar in acetonitril.
Elke tablet bevat als inactieve ingrediënten: lactose, magnesiumstearaat, hydroxypropylmethylcellulose, polyethyleenglycol, povidon, natriumzetmeelglycolaat en titaniumdioxide.
INDICATIES
Adjuvante behandeling
ARIMIDEX is geïndiceerd voor de adjuvante behandeling van postmenopauzale vrouwen met hormoonreceptorpositieve borstkanker in een vroeg stadium.
Eerstelijnsbehandeling
ARIMIDEX 1 mg is geïndiceerd voor de eerstelijnsbehandeling van postmenopauzale vrouwen met hormoonreceptorpositieve of hormoonreceptor onbekende lokaal gevorderde of gemetastaseerde borstkanker.
Tweedelijnsbehandeling
ARIMIDEX 1 mg is geïndiceerd voor de behandeling van gevorderde borstkanker bij postmenopauzale vrouwen met ziekteprogressie na behandeling met tamoxifen. Patiënten met ER-negatieve ziekte en patiënten die niet reageerden op eerdere tamoxifentherapie, reageerden zelden op ARIMIDEX.
DOSERING EN ADMINISTRATIE
Aanbevolen dosis
De dosis ARIMIDEX is één tablet van 1 mg eenmaal daags. Bij patiënten met gevorderde borstkanker moet ARIMIDEX 1 mg worden voortgezet tot tumorprogressie. ARIMIDEX kan met of zonder voedsel worden ingenomen.
Voor adjuvante behandeling van vroege borstkanker bij postmenopauzale vrouwen is de optimale duur van de behandeling niet bekend. In de ATAC-studie werd ARIMIDEX 1 mg gedurende vijf jaar toegediend [zie Klinische studies ].
Er is geen dosisaanpassing nodig voor patiënten met nierinsufficiëntie of voor oudere patiënten [zie: Gebruik bij specifieke populaties ].
Patiënten met leverinsufficiëntie
Er worden geen dosisaanpassingen aanbevolen voor patiënten met een lichte tot matige leverfunctiestoornis. ARIMIDEX is niet onderzocht bij patiënten met een ernstige leverfunctiestoornis [zie: Gebruik in specifieke populaties ].
HOE GELEVERD
Doseringsvormen en sterke punten
De tabletten zijn wit, biconvex, filmomhuld en bevatten 1 mg anastrozol. De tabletten zijn aan de ene kant bedrukt met een logo bestaande uit een letter "A" (hoofdletter) met een pijlpunt bevestigd aan de voet van het verlengde rechterbeen van de "A" en aan de andere kant met de tabletsterkte-markering "Adx 1 ”.
Opslag en behandeling
Deze tabletten worden geleverd in flesjes van 30 tabletten ( NDC 0310-0201-30).
Opslag
Bewaren bij een gecontroleerde kamertemperatuur, 20-25 ° C (68-77 ° F) [zie USP ].
Gedistribueerd door: AstraZeneca Pharmaceuticals LP Wilmington, DE 19850. Herzien: mei 2014
BIJWERKINGEN
Ernstige bijwerkingen van ARIMIDEX die bij minder dan 1 op de 10.000 patiënten optreden, zijn: 1) huidreacties zoals laesies, zweren of blaren; 2) allergische reacties met zwelling van het gezicht, de lippen, de tong en/of de keel. Dit kan problemen met slikken en/of ademen veroorzaken; en 3) veranderingen in bloedtesten van de leverfunctie, waaronder ontsteking van de lever met symptomen die kunnen bestaan uit een algemeen gevoel van onwel zijn, met of zonder geelzucht, leverpijn of leverzwelling.
Vaak voorkomende bijwerkingen (die optreden met een incidentie van ≥ 10%) bij vrouwen die ARIMIDEX 1 mg gebruikten omvatten: opvliegers, asthenie, artritis, pijn, artralgie, hypertensie, depressie, misselijkheid en braken, huiduitslag, osteoporose, fracturen, rugpijn, slapeloosheid, hoofdpijn, botpijn, perifeer oedeem, toegenomen hoest, dyspneu, faryngitis en lymfoedeem.
In de ATAC-studie waren opvliegers de meest voorkomende gemelde bijwerking (> 0,1%) die leidde tot stopzetting van de behandeling voor beide behandelingsgroepen, hoewel er minder patiënten waren die de behandeling staakten als gevolg van opvliegers in de ARIMIDEX 1 mg-groep.
Omdat klinische onderzoeken onder sterk uiteenlopende omstandigheden worden uitgevoerd, kunnen de bijwerkingen die in de klinische onderzoeken van een geneesmiddel zijn waargenomen niet direct worden vergeleken met de percentages in de klinische onderzoeken van een ander geneesmiddel en komen mogelijk niet overeen met de percentages die in de praktijk worden waargenomen.
Ervaring met klinische proeven
Adjuvante therapie
Gegevens over bijwerkingen voor adjuvante therapie zijn gebaseerd op de ATAC-studie [zie: Klinische studies ]. De mediane duur van adjuvante behandeling voor veiligheidsevaluatie was 59,8 maanden en 59,6 maanden voor patiënten die respectievelijk ARIMIDEX 1 mg en tamoxifen 20 mg kregen.
Bijwerkingen die optreden met een incidentie van ten minste 5% in beide behandelingsgroepen tijdens de behandeling of binnen 14 dagen na het einde van de behandeling worden weergegeven in Tabel 1.
Bepaalde bijwerkingen en combinaties van bijwerkingen werden prospectief gespecificeerd voor analyse, op basis van de bekende farmacologische eigenschappen en bijwerkingenprofielen van de twee geneesmiddelen (zie tabel 2).
Ischemische cardiovasculaire gebeurtenissen
Tussen de behandelingsarmen in de totale populatie van 6186 patiënten was er geen statistisch verschil in ischemische cardiovasculaire voorvallen (4% ARIMIDEX vs. 3% tamoxifen).
In de totale populatie werd angina pectoris gemeld bij 71/3092 (2,3%) patiënten in de ARIMIDEX 1 mg-arm en 51/3094 (1,6%) patiënten in de tamoxifen-arm; myocardinfarct werd gemeld bij 37/3092 (1,2%) patiënten in de ARIMIDEX 1 mg-arm en 34/3094 (1,1%) patiënten in de tamoxifen-arm.
Bij vrouwen met een reeds bestaande ischemische hartziekte 465/6186 (7,5%) was de incidentie van ischemische cardiovasculaire voorvallen 17% bij patiënten die ARIMIDEX kregen en 10% bij patiënten die tamoxifen kregen. In deze patiëntenpopulatie werd angina pectoris gemeld bij 25/216 (11,6%) patiënten die ARIMIDEX kregen en bij 13/249 (5,2%) patiënten die tamoxifen kregen; myocardinfarct werd gemeld bij 2/216 (0,9%) patiënten die ARIMIDEX kregen en bij 8/249 (3,2%) patiënten die tamoxifen kregen.
Bevindingen van botmineraaldichtheid
Resultaten van de botsubstudie van de ATAC-studie na 12 en 24 maanden toonden aan dat patiënten die ARIMIDEX 1 mg kregen, een gemiddelde afname hadden van zowel de lumbale wervelkolom als de totale botmineraaldichtheid (BMD) van de heup in vergelijking met baseline. Patiënten die tamoxifen kregen, hadden een gemiddelde toename van zowel de lumbale wervelkolom als de totale heup BMD vergeleken met baseline.
Omdat ARIMIDEX de circulerende oestrogeenspiegels verlaagt, kan het een vermindering van de botmineraaldichtheid veroorzaken.
Een postmarketingonderzoek beoordeelde de gecombineerde effecten van ARIMIDEX 1 mg en het bisfosfonaatrisedronaat op veranderingen ten opzichte van baseline in BMD en markers van botresorptie en -vorming bij postmenopauzale vrouwen met hormoonreceptorpositieve vroege borstkanker. Alle patiënten kregen calcium- en vitamine D-suppletie. Na 12 maanden werden bij patiënten die geen bisfosfonaten kregen, een kleine afname van de botmineraaldichtheid van de lumbale wervelkolom waargenomen. Behandeling met bisfosfonaten behield de botdichtheid bij de meeste patiënten met een risico op fracturen.
Bij postmenopauzale vrouwen met vroege borstkanker die volgens planning met ARIMIDEX 1 mg zullen worden behandeld, moet de botstatus worden behandeld volgens de behandelrichtlijnen die al beschikbaar zijn voor postmenopauzale vrouwen met een vergelijkbaar risico op fragiliteitsfracturen.
cholesterol
Tijdens de ATAC-studie werd gemeld dat meer patiënten die ARIMIDEX 1 mg kregen een verhoogd serumcholesterol hadden dan patiënten die tamoxifen kregen (respectievelijk 9% versus 3,5%).
Een postmarketingonderzoek evalueerde ook alle mogelijke effecten van ARIMIDEX op het lipidenprofiel. In de primaire analysepopulatie voor lipiden (alleen ARIMIDEX 1 mg) was er geen klinisch significante verandering in LDL-C vanaf baseline tot 12 maanden en HDL-C vanaf baseline tot 12 maanden.
In de secundaire populatie voor lipiden (ARIMIDEX+risedronaat) was er ook geen klinisch significante verandering in LDL-C en HDL-C vanaf baseline tot 12 maanden.
In beide populaties voor lipiden was er na 12 maanden geen klinisch significant verschil in totaal cholesterol (TC) of serumtriglyceriden (TG) in vergelijking met baseline.
In deze studie had een behandeling van 12 maanden met alleen ARIMIDEX 1 mg een neutraal effect op het lipidenprofiel. Combinatiebehandeling met ARIMIDEX en risedronaat had ook een neutraal effect op het lipidenprofiel.
De studie levert bewijs dat postmenopauzale vrouwen met vroege borstkanker die volgens planning met ARIMIDEX 1 mg zullen worden behandeld, moeten worden behandeld met behulp van de huidige richtlijnen van het National Cholesterol Education Program voor cardiovasculair risicogebaseerd beheer van individuele patiënten met LDL-verhogingen.
Andere bijwerkingen
Patiënten die ARIMIDEX kregen, hadden een toename van gewrichtsaandoeningen (waaronder artritis, artrose en artralgie) in vergelijking met patiënten die tamoxifen kregen. Patiënten die ARIMIDEX kregen, hadden een toename in de incidentie van alle fracturen (met name fracturen van wervelkolom, heup en pols) [315 (10%)] vergeleken met patiënten die tamoxifen kregen [209 (7%)].
Patiënten die ARIMIDEX 1 mg kregen, hadden een hogere incidentie van carpaaltunnelsyndroom [78 (2,5%)] in vergelijking met patiënten die tamoxifen kregen [22 (0,7%)].
Vaginale bloedingen kwamen vaker voor bij de met tamoxifen behandelde patiënten dan bij de met ARIMIDEX behandelde patiënten 317 (10%) versus 167 (5%).
Patiënten die ARIMIDEX kregen, hadden een lagere incidentie van opvliegers, vaginale bloedingen, vaginale afscheiding, endometriumkanker, veneuze trombo-embolische voorvallen en ischemische cerebrovasculaire voorvallen in vergelijking met patiënten die tamoxifen kregen.
10 jaar mediane follow-up veiligheidsresultaten van het ATAC-onderzoek
De resultaten zijn consistent met de vorige analyses.
Ernstige bijwerkingen waren vergelijkbaar tussen ARIMIDEX (50%) en tamoxifen (51%).
- Cardiovasculaire voorvallen kwamen overeen met de bekende veiligheidsprofielen van ARIMIDEX en tamoxifen.
- De cumulatieve incidenties van alle eerste fracturen (zowel ernstige als niet-ernstige, optredend tijdens of na de behandeling) was hoger in de ARIMIDEX-groep (15%) dan in de tamoxifen-groep (11%). Dit verhoogde percentage eerste fracturen tijdens de behandeling zette zich niet voort in de follow-upperiode na de behandeling.
- De cumulatieve incidentie van nieuwe primaire kankers was vergelijkbaar in de ARIMIDEX 1 mg-groep (13,7%) vergeleken met de tamoxifen-groep (13,9%). In overeenstemming met de eerdere analyses was endometriumkanker hoger in de tamoxifen-groep (0,8%) in vergelijking met de ARIMIDEX-groep (0,2%).
- Het totale aantal sterfgevallen (tijdens of off-trial behandeling) was vergelijkbaar tussen de behandelingsgroepen. Er waren meer sterfgevallen gerelateerd aan borstkanker in de tamoxifen dan in de ARIMIDEX 1 mg behandelingsgroep.
Eerstelijnstherapie
Bijwerkingen die optreden met een incidentie van ten minste 5% in beide behandelingsgroepen van onderzoeken 0030 en 0027 tijdens of binnen 2 weken na het einde van de behandeling worden weergegeven in Tabel 3.
Minder frequente bijwerkingen gemeld bij patiënten die ARIMIDEX 1 mg l mg kregen in Trial 0030 of Trial 0027 waren vergelijkbaar met die gemeld voor tweedelijnstherapie.
Op basis van resultaten van tweedelijnstherapie en het vastgestelde veiligheidsprofiel van tamoxifen, werden de incidenties van 9 vooraf gespecificeerde categorieën van bijwerkingen die mogelijk causaal verband hielden met een of beide therapieën vanwege hun farmacologie, statistisch geanalyseerd. Er werden geen significante verschillen gezien tussen de behandelingsgroepen.
Tweedelijnstherapie
ARIMIDEX 1 mg werd verdragen in twee gecontroleerde klinische onderzoeken (dwz Trials 0004 en 0005), waarbij minder dan 3,3% van de met ARIMIDEX behandelde patiënten en 4,0% van de met megestrolacetaat behandelde patiënten stopten vanwege een bijwerking.
De belangrijkste bijwerking die vaker voorkomt bij ARIMIDEX dan bij megestrolacetaat was diarree. Bijwerkingen die zijn gemeld bij meer dan 5% van de patiënten in een van de behandelingsgroepen in deze twee gecontroleerde klinische onderzoeken, ongeacht de causaliteit, worden hieronder weergegeven:
Lichaam als geheel: griep syndroom; koorts; nek pijn; malaise; accidenteel letsel; infectie
Cardiovasculair: hypertensie; tromboflebitis
lever: Gamma GT verhoogd; SGOT verhoogd; SGPT verhoogd Hematologisch: bloedarmoede; leukopenie
Metabool en voedingswaarde: Alkalische fosfatase verhoogd; gewichtsverlies
De gemiddelde serumconcentraties van totaal cholesterol namen toe met 0, 5 mmol / l bij patiënten die ARIMIDEX kregen. Het is aangetoond dat verhogingen van LDL-cholesterol bijdragen aan deze veranderingen.
Musculoskeletaal: Spierpijn; artralgie; pathologische breuk
Zenuwachtig: Slaperigheid; verwardheid; slapeloosheid; ongerustheid; nervositeit
Ademhaling: sinusitis; bronchitis; rinitis
Huid en aanhangsels: Haaruitval (alopecia); jeuk
Urogenitaal: Urineweginfectie; Borstpijn
De incidenties van de volgende groepen met bijwerkingen die mogelijk causaal verband hielden met een of beide therapieën vanwege hun farmacologie, werden statistisch geanalyseerd: gewichtstoename, oedeem, trombo-embolische aandoeningen, gastro-intestinale stoornissen, opvliegers en vaginale droogheid. Deze zes groepen, en de bijwerkingen die in de groepen waren vastgelegd, werden prospectief gedefinieerd. De resultaten zijn weergegeven in de onderstaande tabel.
Postmarketingervaring
Deze bijwerkingen worden vrijwillig gemeld door een populatie van onbekende grootte. Daarom is het niet altijd mogelijk om hun frequentie betrouwbaar in te schatten of een oorzakelijk verband met blootstelling aan geneesmiddelen vast te stellen. Het volgende is gemeld bij het gebruik van Arimidex na goedkeuring:
- Lever- en galaandoeningen, waaronder verhogingen van alkalische fosfatase, alanineaminotransferase, aspartaataminotransferase, gamma-GT en bilirubine; hepatitis
- Huiduitslag, waaronder gevallen van slijmvliesaandoeningen zoals erythema multiforme en Stevens-Johnson-syndroom
- Gevallen van allergische reacties waaronder angio-oedeem, urticaria en anafylaxie [zie CONTRA-INDICATIES ]
- Myalgie, triggerfinger en hypercalciëmie (met of zonder verhoging van het bijschildklierhormoon)
DRUG-INTERACTIES
Tamoxifen
Gelijktijdige toediening van anastrozol en tamoxifen bij borstkankerpatiënten verminderde de plasmaconcentratie van anastrozol met 27%. De gelijktijdige toediening van anastrozol en tamoxifen had echter geen invloed op de farmacokinetiek van tamoxifen of N-desmethyltamoxifen. Bij een mediane follow-up van 33 maanden vertoonde de combinatie van ARIMIDEX en tamoxifen geen enkel werkzaamheidsvoordeel in vergelijking met tamoxifen bij alle patiënten en evenmin bij de hormoonreceptorpositieve subpopulatie. Deze behandelarm werd uit de studie stopgezet [zie Klinische studies ]. Op basis van klinische en farmacokinetische resultaten van de ATAC-studie mag tamoxifen niet worden toegediend met anastrozol.
Oestrogeen
Oestrogeenbevattende therapieën mogen niet worden gebruikt met ARIMIDEX 1 mg, omdat ze de farmacologische werking ervan kunnen verminderen.
Warfarine
In een onderzoek uitgevoerd bij 16 mannelijke vrijwilligers veranderde anastrozol de blootstelling (zoals gemeten door Cmax en AUC) en antistollingsactiviteit (zoals gemeten door protrombinetijd, geactiveerde partiële tromboplastinetijd en trombinetijd) van zowel R- als S- niet. warfarine.
Cytochroom P450 Op basis van in vitro en in vivo resultaten is het onwaarschijnlijk dat gelijktijdige toediening van ARIMIDEX 1 mg andere geneesmiddelen zal beïnvloeden als gevolg van remming van cytochroom P450 [zie KLINISCHE FARMACOLOGIE ].
WAARSCHUWINGEN
Inbegrepen als onderdeel van de PREVENTIEVE MAATREGELEN sectie.
PREVENTIEVE MAATREGELEN
Ischemische cardiovasculaire gebeurtenissen
Bij vrouwen met een reeds bestaande ischemische hartziekte werd een verhoogde incidentie van ischemische cardiovasculaire voorvallen waargenomen met ARIMIDEX in het ATAC-onderzoek (17% van de patiënten op ARIMIDEX 1 mg en 10% van de patiënten op tamoxifen). Overweeg de risico's en voordelen van behandeling met ARIMIDEX bij patiënten met een reeds bestaande ischemische hartziekte [zie: ONGEWENSTE REACTIES ]
Bot effecten
Resultaten van de botsubstudie van de ATAC-studie na 12 en 24 maanden toonden aan dat patiënten die ARIMIDEX kregen, een gemiddelde afname hadden in zowel de lumbale wervelkolom als de totale botmineraaldichtheid (BMD) van de heup in vergelijking met baseline. Patiënten die tamoxifen kregen, hadden een gemiddelde toename van zowel de lumbale wervelkolom als de totale heup BMD vergeleken met baseline. Overweeg monitoring van de botmineraaldichtheid bij patiënten die worden behandeld met ARIMIDEX [zie: ONGEWENSTE REACTIES ].
cholesterol
Tijdens de ATAC-studie werd gemeld dat meer patiënten die ARIMIDEX 1 mg kregen een verhoogd serumcholesterol hadden dan patiënten die tamoxifen kregen (respectievelijk 9% versus 3,5%) [zie ONGEWENSTE REACTIES ].
Informatie over patiëntbegeleiding
Zien Door de FDA goedgekeurde patiëntetikettering (PATINTINFORMATIE).
Zwangerschap
Patiënten moeten erop worden gewezen dat ARIMIDEX 1 mg schade aan de foetus kan veroorzaken. Ze moeten er ook op worden gewezen dat ARIMIDEX 1 mg niet bestemd is voor gebruik bij premenopauzale vrouwen; daarom moeten ze, als ze zwanger worden, stoppen met het gebruik van ARIMIDEX en onmiddellijk contact opnemen met hun arts.
Allergische (overgevoeligheids) reacties
Patiënten moeten worden geïnformeerd over de mogelijkheid van ernstige allergische reacties met zwelling van het gezicht, de lippen, tong en/of keel (angio-oedeem), wat problemen met slikken en/of ademen kan veroorzaken, en ze moeten onmiddellijk medische hulp inroepen.
Ischemische cardiovasculaire gebeurtenissen
Patiënten met een reeds bestaande ischemische hartziekte moeten worden geïnformeerd dat een verhoogde incidentie van cardiovasculaire voorvallen is waargenomen bij gebruik van ARIMIDEX 1 mg in vergelijking met het gebruik van tamoxifen. Als patiënten nieuwe of verergerende pijn op de borst of kortademigheid hebben, moeten ze onmiddellijk medische hulp inroepen.
Bot effecten
Patiënten moeten worden geïnformeerd dat ARIMIDEX het oestrogeengehalte verlaagt. Dit kan leiden tot een verlies van het mineraalgehalte van botten, wat de botsterkte kan verminderen. Een mogelijk gevolg van een verminderd mineraalgehalte van botten is een toename van het risico op fracturen.
cholesterol
Patiënten moeten worden geïnformeerd dat een verhoogd cholesterolgehalte kan worden waargenomen tijdens het gebruik van ARIMIDEX.
Kietelen, tintelingen of gevoelloosheid
Patiënten moeten worden geïnformeerd dat als ze kietelen, tintelingen of gevoelloosheid ervaren, ze hun zorgverlener op de hoogte moeten stellen.
Tamoxifen
Patiënten dienen te worden geadviseerd ARIMIDEX niet samen met Tamoxifen in te nemen.
Gemiste doses
Informeer patiënten dat als ze een dosis overslaan, ze deze innemen zodra ze eraan denken. Als het bijna tijd is voor hun volgende dosis, sla dan de gemiste dosis over en neem de volgende geplande dosis. Patiënten mogen geen twee doses tegelijk innemen.
Niet-klinische toxicologie
Carcinogenese, mutagenese, verminderde vruchtbaarheid
Een conventioneel carcinogenese-onderzoek bij ratten met doses van 1,0 tot 25 mg/kg/dag (ongeveer 10 tot 243 maal de maximaal aanbevolen dagelijkse dosis voor de mens op basis van mg/m²), toegediend via orale sondevoeding gedurende maximaal 2 jaar, onthulde een toename van de incidentie van hepatocellulair adenoom en carcinoom en uteriene stromale poliepen bij vrouwen en schildklieradenoom bij mannen bij de hoge dosis. Een dosisgerelateerde toename werd waargenomen in de incidentie van ovarium- en baarmoederhyperplasie bij vrouwen. Bij 25 mg/kg/dag waren de plasma-AUC0-24 uur-spiegels bij ratten 110 tot 125 keer hoger dan de spiegel die werd vertoond bij postmenopauzale vrijwilligers bij de aanbevolen dosis. Een afzonderlijk carcinogeniteitsonderzoek bij muizen bij orale doses van 5 tot 50 mg/kg/dag (ongeveer 24 tot 243 maal de maximaal aanbevolen dagelijkse dosis voor de mens op basis van mg/m²) gedurende maximaal 2 jaar leidde tot een toename van de incidentie van goedaardige ovariële stromale, epitheliale en granulosaceltumoren op alle dosisniveaus. Een dosisgerelateerde toename van de incidentie van ovariële hyperplasie werd ook waargenomen bij vrouwelijke muizen. Deze ovariële veranderingen worden beschouwd als knaagdierspecifieke effecten van aromatase-remming en zijn van twijfelachtige betekenis voor de mens. De incidentie van lymfosarcoom was verhoogd bij mannen en vrouwen bij de hoge dosis. Bij 50 mg/kg/dag waren de plasma-AUC-spiegels bij muizen 35 tot 40 keer hoger dan de spiegel die werd vertoond bij postmenopauzale vrijwilligers bij de aanbevolen dosis.
Het is niet aangetoond dat ARIMIDEX 1 mg mutageen is in in vitro tests (Ames- en E. coli-bacterietests, CHO-K1-genmutatietest) of clastogeen, noch in vitro (chromosoomafwijkingen in menselijke lymfocyten) of in vivo (micronucleustest bij ratten) .
Orale toediening van anastrozol aan vrouwelijke ratten (vanaf 2 weken voor de paring tot dag 7 van de dracht) veroorzaakte een significante incidentie van onvruchtbaarheid en verminderde het aantal levensvatbare zwangerschappen bij 1 mg/kg/dag (ongeveer 10 maal de aanbevolen dosis voor de mens op basis van mg/m²). en 9 keer hoger dan de AUC0-24 uur gevonden bij postmenopauzale vrijwilligers bij de aanbevolen dosis). Pre-implantatieverlies van eicellen of foetus was verhoogd bij doses gelijk aan of hoger dan 0,02 mg/kg/dag (ongeveer een vijfde van de aanbevolen dosis voor de mens op basis van mg/m²). Herstel van de vruchtbaarheid werd waargenomen na een niet-doseringsperiode van 5 weken die volgde op een dosering van 3 weken. Het is niet bekend of deze effecten die zijn waargenomen bij vrouwelijke ratten wijzen op verminderde vruchtbaarheid bij mensen.
Onderzoeken met meervoudige doses bij ratten die gedurende 6 maanden anastrozol kregen toegediend in doses gelijk aan of hoger dan 1 mg/kg/dag (die plasma-anastrozol Cssmax en AUC 0-24 uur produceerden die 19 en 9 keer hoger waren dan de respectieve waarden gevonden in postmenopauzale vrijwilligers in de aanbevolen dosis) resulteerden in hypertrofie van de eierstokken en de aanwezigheid van folliculaire cysten. Daarnaast werden hyperplastische baarmoeders waargenomen in 6 maanden durende onderzoeken bij vrouwelijke honden die doses kregen die gelijk waren aan of groter dan 1 mg/kg/dag (die plasma-anastrozol Cssmax en AUC0-24 uur produceerden die 22 keer en 16 keer hoger waren dan de respectieve waarden gevonden bij postmenopauzale vrouwen bij de aanbevolen dosis). Het is niet bekend of deze effecten op de voortplantingsorganen van dieren geassocieerd zijn met verminderde vruchtbaarheid bij premenopauzale vrouwen.
Gebruik bij specifieke populaties
Zwangerschap
Zwangerschap Categorie X [zien CONTRA-INDICATIES ]
ARIMIDEX kan schade aan de foetus veroorzaken wanneer het wordt toegediend aan een zwangere vrouw en biedt geen klinisch voordeel voor premenopauzale vrouwen met borstkanker. ARIMIDEX is gecontra-indiceerd bij vrouwen die zwanger zijn of kunnen worden. In dierstudies veroorzaakte anastrozol zwangerschapsfalen, toegenomen zwangerschapsverlies en tekenen van vertraagde ontwikkeling van de foetus. Er zijn geen onderzoeken naar het gebruik van ARIMIDEX 1 mg bij zwangere vrouwen. Als ARIMIDEX 1 mg tijdens de zwangerschap wordt gebruikt, of als de patiënte zwanger wordt terwijl ze dit geneesmiddel krijgt, moet de patiënt op de hoogte zijn van het mogelijke gevaar voor de foetus en het mogelijke risico op zwangerschapsverlies.
In reproductiestudies bij dieren kregen zwangere ratten en konijnen anastrozol tijdens de organogenese in doses gelijk aan of groter dan 1 (ratten) en 1/3 (konijnen) de aanbevolen dosis voor de mens op basis van mg/m². Bij beide soorten passeerde anastrozol de placenta en was er meer zwangerschapsverlies (verhoogd pre- en/of post-implantatieverlies, verhoogde resorptie en verminderd aantal levende foetussen). Bij ratten waren deze effecten dosisgerelateerd en was het gewicht van de placenta significant verhoogd. Foetotoxiciteit, waaronder vertraagde foetale ontwikkeling (dwz onvolledige ossificatie en verlaagd foetaal lichaamsgewicht), trad op bij ratten bij doses anastrozol die piekplasmaspiegels produceerden die 19 keer hoger waren dan serumspiegels bij mensen bij de therapeutische dosis (AUC 0-24 uur 9 keer hoger) . Bij konijnen veroorzaakte anastrozol zwangerschapsfalen bij doses gelijk aan of groter dan 16 maal de aanbevolen dosis voor de mens op basis van mg/m² [zie Diertoxicologie en/of farmacologie ].
Moeders die borstvoeding geven
Het is niet bekend of anastrozol wordt uitgescheiden in de moedermelk. Omdat veel geneesmiddelen worden uitgescheiden in de moedermelk en vanwege de tumorigeniciteit die is aangetoond voor anastrozol in dierstudies, of vanwege de mogelijkheid van ernstige bijwerkingen bij zuigelingen die borstvoeding geven, moet worden besloten of de borstvoeding moet worden gestaakt of dat het geneesmiddel moet worden gestaakt, rekening houdend met de belang van het medicijn voor de moeder.
Pediatrisch gebruik
Klinische onderzoeken bij pediatrische patiënten omvatten een placebogecontroleerd onderzoek bij puberale jongens in de adolescentieleeftijd met gynaecomastie en een eenarmig onderzoek bij meisjes met het McCune-Albright-syndroom en progressieve vroegtijdige puberteit. De werkzaamheid van ARIMIDEX 1 mg bij de behandeling van puberale gynaecomastie bij adolescente jongens en bij de behandeling van vroegtijdige puberteit bij meisjes met het McCune-Albright-syndroom is niet aangetoond.
Gynaecomastie studie
Een gerandomiseerde, dubbelblinde, placebo-gecontroleerde, multi-center studie omvatte 80 jongens met puberale gynaecomastie in de leeftijd van 11 tot 18 jaar. Patiënten werden gerandomiseerd naar een dagelijks regime van ARIMIDEX 1 mg of placebo. Na 6 maanden behandeling was er geen statistisch significant verschil in het percentage patiënten dat een ≥ 50% afname van gynaecomastie ervoer (primaire werkzaamheidsanalyse). Secundaire werkzaamheidsanalyses (absolute verandering in borstvolume, het percentage patiënten met een vermindering van het berekende volume van gynaecomastie, verdwijnen van borstpijn) waren consistent met de primaire werkzaamheidsanalyse. De serumoestradiolconcentraties in maand 6 van de behandeling waren met 15,4% verlaagd in de ARIMIDEX-groep en met 4,5% in de placebogroep.
Bijwerkingen die door de onderzoekers werden beoordeeld als behandelingsgerelateerd, kwamen voor bij 16,3% van de met ARIMIDEX behandelde patiënten en 8,1% van de met placebo behandelde patiënten, met als meest voorkomende acne (7% ARIMIDEX en 2,7% placebo) en hoofdpijn (7 % ARIMIDEX 1 mg en 0% placebo); alle andere bijwerkingen vertoonden kleine verschillen tussen de behandelingsgroepen. Eén patiënt die werd behandeld met ARIMIDEX 1 mg stopte met het onderzoek vanwege een vergroting van de testikels. De gemiddelde verandering in het testikelvolume na 6 maanden behandeling bij baseline was + 6,6 ± 7,9 cm³ bij de met ARIMIDEX behandelde patiënten en + 5,2 ± 8,0 cm³ in de placebogroep.
Onderzoek naar McCune-Albright-syndroom
Een multicenter, eenarmig, open-label onderzoek werd uitgevoerd bij 28 meisjes met het McCune-Albright-syndroom en progressieve vroegtijdige puberteit in de leeftijd van 2 tot
Vijf patiënten (18%) ondervonden bijwerkingen die mogelijk verband hielden met ARIMIDEX. Dit waren misselijkheid, acne, pijn in een extremiteit, verhoogd alaninetransaminase en aspartaattransaminase en allergische dermatitis.
Farmacokinetiek bij pediatrische patiënten
Na meervoudige toediening van 1 mg eenmaal daags bij pediatrische patiënten was de gemiddelde tijd om de maximale anastrozolconcentratie te bereiken 1 uur. De gemiddelde (bereik)dispositieparameters van anastrozol bij pediatrische patiënten werden beschreven door een CL/F van 1,54 l/u (0,77-4,53 l/u) en een V/F van 98,4 l (50,7-330,0 l). De terminale eliminatiehalfwaardetijd was 46,8 uur, wat vergelijkbaar was met de eliminatiehalfwaardetijd die werd waargenomen bij postmenopauzale vrouwen die werden behandeld met anastrozol voor borstkanker. Op basis van een populatiefarmacokinetische analyse was de farmacokinetiek van anastrozol vergelijkbaar bij jongens met puberale gynaecomastie en meisjes met het McCune-Albright-syndroom.
Geriatrisch gebruik
In onderzoeken 0030 en 0027 was ongeveer 50% van de patiënten 65 jaar of ouder. Patiënten ≥ 65 jaar hadden een matig betere tumorrespons en tijd tot tumorprogressie dan patiënten
In de ATAC-studie was 45% van de patiënten 65 jaar of ouder. De werkzaamheid van ARIMIDEX 1 mg vergeleken met tamoxifen bij patiënten van 65 jaar of ouder (N=1413 voor ARIMIDEX en N=1410 voor tamoxifen, de hazard ratio voor ziektevrije overleving was 0,93 [95% BI: 0,80, 1,08]) was minder dan de werkzaamheid waargenomen bij patiënten jonger dan 65 jaar (N=1712 voor ARIMIDEX en N=1706 voor tamoxifen, de hazard ratio voor ziektevrije overleving was 0,79 [95% BI: 0,67; 0,94]).
De farmacokinetiek van anastrozol wordt niet beïnvloed door leeftijd.
Nierfunctiestoornis
Aangezien slechts ongeveer 10% van anastrozol onveranderd in de urine wordt uitgescheiden, heeft de nierfunctiestoornis geen invloed op de totale lichaamsklaring. Dosisaanpassing bij patiënten met nierinsufficiëntie is niet nodig [zie: DOSERING EN ADMINISTRATIE en KLINISCHE FARMACOLOGIE ].
Leverfunctiestoornis
De plasmaconcentraties van anastrozol bij de proefpersonen met levercirrose lagen binnen het bereik van de concentraties die werden waargenomen bij normale proefpersonen in alle klinische onderzoeken. Daarom is een dosisaanpassing ook niet nodig bij patiënten met stabiele levercirrose. ARIMIDEX is niet onderzocht bij patiënten met een ernstige leverfunctiestoornis [zie: DOSERING EN ADMINISTRATIE en KLINISCHE FARMACOLOGIE ].
OVERDOSERING
Er zijn klinische onderzoeken uitgevoerd met ARIMIDEX, tot 60 mg in een enkele dosis gegeven aan gezonde mannelijke vrijwilligers en tot 10 mg per dag gegeven aan postmenopauzale vrouwen met gevorderde borstkanker; deze doseringen werden getolereerd. Een enkele dosis ARIMIDEX 1 mg die levensbedreigende symptomen veroorzaakt, is niet vastgesteld. Er is geen specifiek antidotum voor overdosering en de behandeling moet symptomatisch zijn. Houd er bij de behandeling van een overdosis rekening mee dat er mogelijk meerdere middelen zijn ingenomen. Braken kan worden opgewekt als de patiënt alert is. Dialyse kan nuttig zijn omdat ARIMIDEX niet sterk eiwitgebonden is. Algemene ondersteunende zorg, waaronder frequente controle van de vitale functies en nauwkeurige observatie van de patiënt, is geïndiceerd.
CONTRA-INDICATIES
Zwangerschap en premenopauzale vrouwen
ARIMIDEX 1 mg kan schade aan de foetus veroorzaken wanneer het wordt toegediend aan een zwangere vrouw en biedt geen klinisch voordeel voor premenopauzale vrouwen met borstkanker. ARIMIDEX 1 mg is gecontra-indiceerd bij vrouwen die zwanger zijn of kunnen worden. Er zijn geen adequate en goed gecontroleerde onderzoeken bij zwangere vrouwen die ARIMIDEX gebruiken. Als ARIMIDEX 1 mg tijdens de zwangerschap wordt gebruikt, of als de patiënte zwanger wordt tijdens het gebruik van dit geneesmiddel, moet de patiënt op de hoogte worden gesteld van het mogelijke gevaar voor een foetus of het mogelijke risico op verlies van de zwangerschap [zie Gebruik in specifieke populaties ].
overgevoeligheid
ARIMIDEX 1 mg is gecontra-indiceerd bij elke patiënt die een overgevoeligheidsreactie heeft vertoond op het geneesmiddel of op een van de hulpstoffen. Waargenomen reacties zijn onder meer anafylaxie, angio-oedeem en urticaria [zie: ONGEWENSTE REACTIES ]
KLINISCHE FARMACOLOGIE
Werkingsmechanisme
De groei van veel kankers van de borst wordt gestimuleerd of in stand gehouden door oestrogenen.
Bij postmenopauzale vrouwen worden oestrogenen voornamelijk afgeleid van de werking van het aromatase-enzym, dat bijnierandrogenen (voornamelijk androstenedion en testosteron) omzet in oestron en estradiol. De onderdrukking van oestrogeenbiosynthese in perifere weefsels en in het kankerweefsel zelf kan daarom worden bereikt door specifiek het aromatase-enzym te remmen.
Anastrozol is een selectieve niet-steroïde aromataseremmer. Het verlaagt de serumestradiolconcentraties aanzienlijk en heeft geen waarneembaar effect op de vorming van bijniercorticosteroïden of aldosteron.
farmacodynamiek
Effect op oestradiol
Gemiddelde serumconcentraties van estradiol werden geëvalueerd in meerdere dagelijkse doseringsonderzoeken met 0,5, 1, 3, 5 en 10 mg ARIMIDEX bij postmenopauzale vrouwen met gevorderde borstkanker. Klinisch significante onderdrukking van serumestradiol werd bij alle doses waargenomen. Doses van 1 mg en hoger resulteerden in onderdrukking van de gemiddelde serumconcentraties van estradiol tot de ondergrens van detectie (3,7 pmol/l). De aanbevolen dagelijkse dosis, ARIMIDEX 1 mg, verminderde oestradiol met ongeveer 70% binnen 24 uur en met ongeveer 80% na 14 dagen dagelijkse dosering. De onderdrukking van serumoestradiol bleef tot 6 dagen na stopzetting van de dagelijkse dosering met ARIMIDEX 1 mg gehandhaafd.
Het effect van ARIMIDEX 1 mg bij premenopauzale vrouwen met vroege of gevorderde borstkanker is niet onderzocht. Omdat aromatisering van bijnierandrogenen geen significante bron van estradiol is bij premenopauzale vrouwen, wordt niet verwacht dat ARIMIDEX de estradiolspiegels bij premenopauzale vrouwen verlaagt.
Effect op corticosteroïden
In meerdere dagelijkse doseringsonderzoeken met 3, 5 en 10 mg werd de selectiviteit van anastrozol beoordeeld door de effecten op de synthese van corticosteroïden te onderzoeken. Voor alle doses had anastrozol geen invloed op de secretie van cortisol of aldosteron bij baseline of als reactie op ACTH. Bij anastrozol is geen substitutietherapie met glucocorticoïden of mineralocorticoïden nodig.
Andere endocriene effecten
In meerdere dagelijkse doseringsproeven met 5 en 10 mg werd thyroïdstimulerend hormoon (TSH) gemeten; er was geen verhoging van TSH tijdens de toediening van ARIMIDEX. ARIMIDEX bezit geen directe progestogene, androgene of oestrogene activiteit bij dieren, maar verstoort de circulerende niveaus van progesteron, androgenen en oestrogenen.
Farmacokinetiek
Absorptie
Remming van aromatase-activiteit is voornamelijk te wijten aan anastrozol, het moedergeneesmiddel. Absorptie van anastrozol is snel en maximale plasmaconcentraties treden doorgaans op binnen 2 uur na toediening in nuchtere toestand. Studies met radioactief gelabelde geneesmiddelen hebben aangetoond dat oraal toegediend anastrozol goed in de systemische circulatie wordt opgenomen. Voedsel vermindert de snelheid, maar niet de algehele mate van absorptie van anastrozol. De gemiddelde Cmax van anastrozol nam af met 16% en de mediane Tmax was vertraagd van 2 tot 5 uur wanneer anastrozol 30 minuten na voedsel werd toegediend. De farmacokinetiek van anastrozol is lineair over het dosisbereik van 1 tot 20 mg en verandert niet bij herhaalde dosering. De farmacokinetiek van anastrozol was vergelijkbaar bij patiënten en gezonde vrijwilligers.
Verdeling
Steady-state plasmaspiegels zijn ongeveer 3 tot 4 keer hoger dan de spiegels die worden waargenomen na een enkele dosis ARIMIDEX. De plasmaconcentraties benaderen de steady-state niveaus bij een eenmaal daagse dosering van ongeveer 7 dagen. Anastrozol is voor 40% gebonden aan plasma-eiwitten in het therapeutische bereik.
Metabolisme
Metabolisme van anastrozol vindt plaats door N-dealkylering, hydroxylering en glucuronidering. Drie metabolieten van anastrozol (triazool, een glucuronideconjugaat van hydroxy-anastrozol en een glucuronideconjugaat van anastrozol zelf) zijn geïdentificeerd in menselijk plasma en urine. De belangrijkste circulerende metaboliet van anastrozol, triazol, heeft geen farmacologische activiteit.
Anastrozol remde in vitro reacties gekatalyseerd door cytochroom P450 1A2, 2C8/9 en 3A4 met Ki-waarden die ongeveer 30 keer hoger waren dan de gemiddelde steady-state Cmax-waarden die werden waargenomen na een dagelijkse dosis van 1 mg. Anastrozol had in vitro geen remmend effect op reacties gekatalyseerd door cytochroom P450 2A6 of 2D6. Toediening van een enkelvoudige dosis van 30 mg/kg of meerdere doses van 10 mg/kg anastrozol aan gezonde proefpersonen had geen effect op de klaring van antipyrine of op de urinewinning van antipyrinemetabolieten.
uitscheiding
Vijfentachtig procent van het radioactief gelabelde anastrozol werd teruggevonden in de ontlasting en urine. Het levermetabolisme is verantwoordelijk voor ongeveer 85% van de eliminatie van anastrozol. De renale eliminatie is goed voor ongeveer 10% van de totale klaring. De gemiddelde eliminatiehalfwaardetijd van anastrozol is 50 uur.
Effect van geslacht en leeftijd
De farmacokinetiek van anastrozol is onderzocht bij postmenopauzale vrouwelijke vrijwilligers en patiënten met borstkanker. Er werden geen leeftijdgerelateerde effecten waargenomen in het bereik 80 jaar.
Effect van ras
De serumspiegels van oestradiol en oestronsulfaat waren vergelijkbaar tussen Japanse en blanke postmenopauzale vrouwen die gedurende 16 dagen dagelijks 1 mg anastrozol kregen. Anastrozol gemiddelde steady-state minimale plasmaconcentraties bij blanke en Japanse postmenopauzale vrouwen waren respectievelijk 25,7 en 30,4 ng/ml.
Effect van nierinsufficiëntie
De farmacokinetiek van anastrozol is onderzocht bij proefpersonen met een nierfunctiestoornis. De renale klaring van anastrozol nam proportioneel af met de creatinineklaring en was ongeveer 50% lager bij vrijwilligers met een ernstige nierfunctiestoornis (creatinineklaring DOSERING EN ADMINISTRATIE en Gebruik in specifieke populaties ].
Effect van leverfunctiestoornis
De farmacokinetiek van anastrozol is onderzocht bij personen met levercirrose gerelateerd aan alcoholmisbruik. De schijnbare orale klaring (CL/F) van anastrozol was ongeveer 30% lager bij proefpersonen met stabiele levercirrose dan bij controlepersonen met een normale leverfunctie. Deze plasmaconcentraties lagen echter nog steeds binnen het bereik van waarden die werden waargenomen bij normale proefpersonen. Het effect van ernstige leverinsufficiëntie is niet onderzocht. Er is geen dosisaanpassing nodig voor stabiele levercirrose [zie DOSERING EN ADMINISTRATIE en Gebruik bij specifieke populaties ].
Dierlijke toxicologie en/of farmacologie
Reproductieve toxicologie
Er is gevonden dat anastrozol de placenta passeert na orale toediening van 0,1 mg/kg bij ratten en konijnen (respectievelijk ongeveer 1 en 1,9 maal de aanbevolen dosis voor de mens, op basis van mg/m²). Studies bij zowel ratten als konijnen met doses gelijk aan of groter dan respectievelijk 0,1 en 0,02 mg/kg/dag (respectievelijk ongeveer 1 en 1/3 van de aanbevolen dosis voor de mens op basis van mg/m²), toegediend tijdens de periode van organogenese toonde aan dat anastrozol het zwangerschapsverlies verhoogde (verhoogd pre- en/of post-implantatieverlies, verhoogde resorptie en verminderd aantal levende foetussen); effecten waren dosisgerelateerd bij ratten. Het gewicht van de placenta was significant verhoogd bij ratten bij doses van 0,1 mg/kg/dag of meer.
Bewijs van foetotoxiciteit, waaronder vertraagde foetale ontwikkeling (dwz onvolledige ossificatie en verlaagd foetaal lichaamsgewicht), werd waargenomen bij ratten die doses van 1 mg/kg/dag kregen toegediend (die plasma-anastrozol Cssmax en AUC 0-24 uur produceerden die 19 keer en 9 keer hoger dan de respectieve waarden gevonden bij postmenopauzale vrijwilligers bij de aanbevolen dosis). Er was geen bewijs van teratogeniteit bij ratten die doses tot 1,0 mg/kg/dag kregen toegediend. Bij konijnen veroorzaakte anastrozol zwangerschapsfalen bij doses gelijk aan of hoger dan 1,0 mg/kg/dag (ongeveer 16 maal de aanbevolen dosis voor de mens op basis van mg/m²); er was geen bewijs van teratogeniteit bij konijnen die 0,2 mg/kg/dag kregen (ongeveer 3 maal de aanbevolen dosis voor de mens op basis van mg/m²).
Klinische studies
Adjuvante behandeling van borstkanker bij postmenopauzale vrouwen
In een dubbelblinde multicenterstudie (ATAC) werden 9.366 postmenopauzale vrouwen met operabele borstkanker gerandomiseerd naar adjuvante behandeling met ARIMIDEX 1 mg per dag, tamoxifen 20 mg per dag, of een combinatie van de twee behandelingen gedurende vijf jaar of tot terugkeer van de ziekte.
Het primaire eindpunt van de studie was ziektevrije overleving (dwz tijd tot optreden van een recidief op afstand of lokaal, of contralaterale borstkanker of overlijden door welke oorzaak dan ook). Secundaire eindpunten van de studie waren ziektevrije overleving op afstand, de incidentie van contralaterale borstkanker en totale overleving. Bij een mediane follow-up van 33 maanden liet de combinatie van ARIMIDEX en tamoxifen geen enkel werkzaamheidsvoordeel zien in vergelijking met tamoxifen bij alle patiënten en bij de hormoonreceptor-positieve subpopulatie. Deze behandelarm werd uit de studie gehaald. Op basis van klinische en farmacokinetische resultaten van het ATAC-onderzoek mag tamoxifen niet worden toegediend met anastrozol [zie DRUG-INTERACTIES ].
Demografische en andere baselinekenmerken waren vergelijkbaar tussen de drie behandelingsgroepen (zie tabel 7).
Patiënten in de twee monotherapie-armen van de ATAC-studie werden gedurende een mediane periode van 60 maanden (5 jaar) behandeld en gevolgd gedurende een mediane periode van 68 maanden. De ziektevrije overleving in de intent-to-treat-populatie was statistisch significant verbeterd [Hazard Ratio (HR) = 0,87, 95% BI: 0,78, 0,97, p=0,0127] in de ARIMIDEX-arm vergeleken met de tamoxifen-arm. In de hormoonreceptor-positieve subpopulatie die ongeveer 84% van de proefpatiënten vertegenwoordigde, was de ziektevrije overleving ook statistisch significant verbeterd (HR = 0,83, 95% BI: 0,73, 0,94, p=0,0049) in de ARIMIDEX 1 mg-arm vergeleken met de tamoxifen arm.
Figuur 1: Ziektevrije overleving Kaplan Meier overlevingscurve voor alle patiënten gerandomiseerd naar ARIMIDEX 1 mg of tamoxifen monotherapie in de ATAC-studie (Intent-to-Treat) 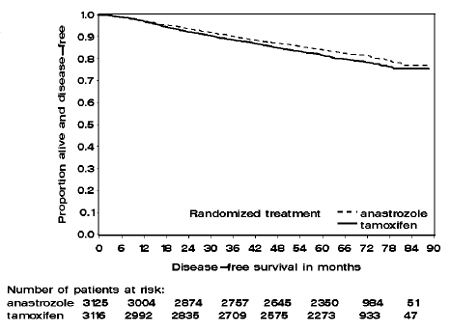
Figuur 2: Ziektevrije overleving voor hormoonreceptor-positieve subpopulatie van patiënten gerandomiseerd naar ARIMIDEX of tamoxifen monotherapie in het ATAC-onderzoek 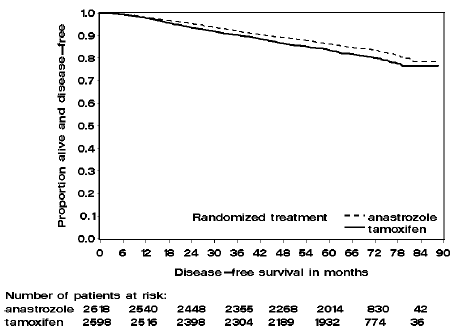
De overlevingsgegevens met een follow-up van 68 maanden zijn weergegeven in Tabel 9.
In de groep patiënten die eerder adjuvante chemotherapie hadden ondergaan (N=698 voor ARIMIDEX 1 mg en N=647 voor tamoxifen), was de hazard ratio voor ziektevrije overleving 0,91 (95% BI: 0,73 tot 1,13) in de ARIMIDEX-arm vergeleken met de tamoxifen-arm.
De frequentie van individuele gebeurtenissen in de intent-to-treat-populatie en de hormoonreceptor-positieve subpopulatie wordt beschreven in Tabel 8.
Een samenvatting van de resultaten van de studiewerkzaamheid wordt gegeven in Tabel 9.
10 jaar mediane follow-up Werkzaamheidsresultaten van het ATAC-onderzoek
In een daaropvolgende analyse van de ATAC-studie werden patiënten in de twee monotherapiearmen gevolgd gedurende een mediane periode van 120 maanden (10 jaar). De patiënten kregen de onderzoeksbehandeling gedurende een mediane periode van 60 maanden (5 jaar) (zie tabel 10).
Figuur 3: Ziektevrije overleving Kaplan Meier overlevingscurve voor alle patiënten gerandomiseerd naar ARIMIDEX of tamoxifen monotherapie in het ATAC-onderzoek (Intent-to-Treat)a 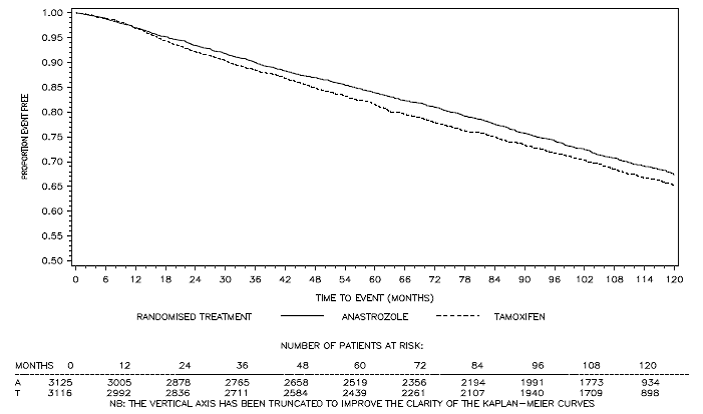
a Het aandeel patiënten met een follow-up van 120 maanden was 29,4%.
Figuur 4: Ziektevrije overleving voor hormoonreceptor-positieve subpopulatie van patiënten gerandomiseerd naar ARIMIDEX 1 mg of tamoxifen monotherapie in het ATAC-onderzoek 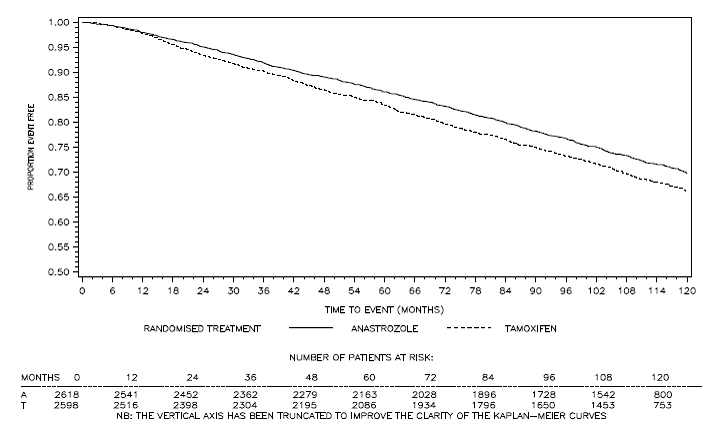
bHet aandeel patiënten met een follow-up van 120 maanden was 29,8%.
Eerstelijnstherapie bij postmenopauzale vrouwen met gevorderde borstkanker
Er zijn twee dubbelblinde, gecontroleerde klinische onderzoeken met een vergelijkbare opzet (0030, een Noord-Amerikaanse studie en 0027, een overwegend Europese studie) uitgevoerd om de werkzaamheid van ARIMIDEX 1 mg te beoordelen in vergelijking met tamoxifen als eerstelijnstherapie voor hormoonreceptorpositieve of hormoonreceptoren. onbekende lokaal gevorderde of gemetastaseerde borstkanker bij postmenopauzale vrouwen. In totaal werden 1021 patiënten tussen de 30 en 92 jaar oud gerandomiseerd om een proefbehandeling te krijgen. Patiënten werden gerandomiseerd om 1 mg ARIMIDEX 1 mg eenmaal daags of 20 mg tamoxifen eenmaal daags te krijgen. De primaire eindpunten voor beide onderzoeken waren tijd tot tumorprogressie, objectieve tumorrespons en veiligheid.
Demografische kenmerken en andere baselinekenmerken, waaronder patiënten met meetbare en geen meetbare ziekte, patiënten die eerdere adjuvante therapie kregen, de plaats van gemetastaseerde ziekte en etnische afkomst waren vergelijkbaar voor de twee behandelingsgroepen voor beide onderzoeken. De volgende tabel geeft een samenvatting van de hormoonreceptorstatus bij binnenkomst voor alle gerandomiseerde patiënten in onderzoeken 0030 en 0027.
Voor de primaire eindpunten toonde onderzoek 0030 aan dat ARIMIDEX een statistisch significant voordeel had ten opzichte van tamoxifen (p=0,006) voor wat betreft de tijd tot tumorprogressie; objectieve tumorresponspercentages waren vergelijkbaar voor ARIMIDEX 1 mg en tamoxifen. Proef 0027 toonde aan dat ARIMIDEX 1 mg en tamoxifen vergelijkbare objectieve tumorresponspercentages en tijd tot tumorprogressie hadden (zie Tabel 12 en Figuren 5 en 6).
Tabel 12 hieronder geeft een samenvatting van de resultaten van onderzoek 0030 en onderzoek 0027 voor de primaire werkzaamheidseindpunten.
Figuur 5: Kaplan-Meier-waarschijnlijkheid van tijd tot ziekteprogressie voor alle gerandomiseerde patiënten (intent-to-treat) in Trial 0030 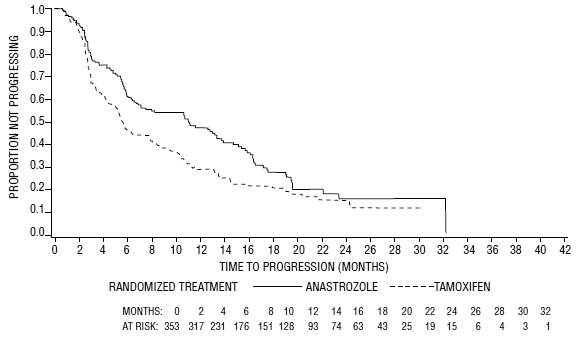
Figuur 6: Kaplan-Meier-waarschijnlijkheid van tijd tot progressie voor alle gerandomiseerde patiënten (intent-to-treat) in Trial 0027 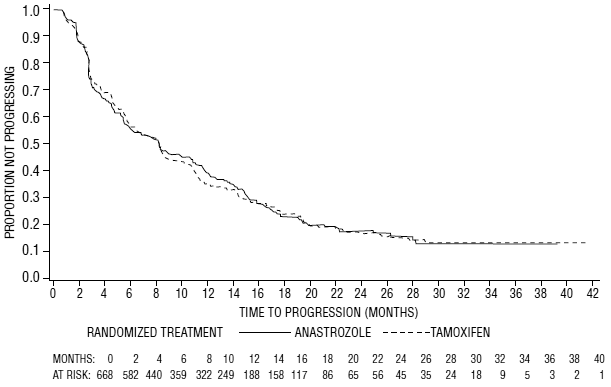
Resultaten van de secundaire eindpunten ondersteunden de resultaten van de primaire werkzaamheidseindpunten. Er waren te weinig sterfgevallen in de behandelingsgroepen van beide onderzoeken om conclusies te trekken over de algehele overlevingsverschillen.
Tweedelijnstherapie bij postmenopauzale vrouwen met gevorderde borstkanker die ziekteprogressie hadden na tamoxifentherapie
Anastrozol is onderzocht in twee gecontroleerde klinische onderzoeken (0004, een Noord-Amerikaans onderzoek; 0005, een overwegend Europees onderzoek) bij postmenopauzale vrouwen met gevorderde borstkanker die ziekteprogressie vertoonden na behandeling met tamoxifen voor gevorderde of vroege borstkanker. Sommige patiënten hadden ook eerder een cytotoxische behandeling ondergaan. De meeste patiënten waren ER-positief; een kleinere fractie was ER-onbekend of ER-negatief; de ER-negatieve patiënten kwamen alleen in aanmerking als ze een positieve reactie op tamoxifen hadden gehad. Geschikte patiënten met meetbare en niet-meetbare ziekte werden gerandomiseerd om ofwel een enkele dagelijkse dosis van 1 mg of 10 mg ARIMIDEX of megestrolacetaat 40 mg viermaal daags te krijgen. De onderzoeken waren dubbelblind met betrekking tot ARIMIDEX. Tijd tot progressie en objectieve respons (alleen patiënten met meetbare ziekte konden als partiële responders worden beschouwd) waren de primaire werkzaamheidsvariabelen. Objectieve responspercentages werden berekend op basis van de criteria van de Union Internationale Contre le Cancer (UICC). De snelheid van langdurige (meer dan 24 weken) stabiele ziekte, de snelheid van progressie en overleving werden ook berekend.
Beide onderzoeken omvatten meer dan 375 patiënten; demografische gegevens en andere baselinekenmerken waren vergelijkbaar voor de drie behandelingsgroepen in elk onderzoek. Patiënten in de 0005-studie hadden beter gereageerd op eerdere behandeling met tamoxifen. Van de patiënten die deelnamen aan een eerdere behandeling met tamoxifen voor gevorderde ziekte (58% in Trial 0004; 57% in Trial 0005), meldde de primaire onderzoeker 18% van deze patiënten in Trial 0004 en 42% in Trial 0005 te hebben gereageerd. In Trial 0004 was 81% van de patiënten ER-positief, 13% was ER-onbekend en 6% was ER-negatief. In Trial 0005 was 58% van de patiënten ER-positief, 37% was ER-onbekend en 5% was ER-negatief. In Trial 0004 had 62% van de patiënten een meetbare ziekte vergeleken met 79% in Trial 0005. De plaatsen van gemetastaseerde ziekte waren vergelijkbaar tussen de behandelingsgroepen voor elke trial. Gemiddeld had 40% van de patiënten metastasen in de weke delen; 60% had botmetastasen; en 40% had viscerale (15% lever) metastasen.
De werkzaamheidsresultaten van de twee onderzoeken waren vergelijkbaar, zoals weergegeven in tabel 13. In beide onderzoeken waren er geen significante verschillen tussen de behandelingsarmen met betrekking tot de werkzaamheidsparameters die in de onderstaande tabel worden vermeld.
Wanneer gegevens van de twee gecontroleerde onderzoeken worden samengevoegd, waren de objectieve responspercentages en mediane tijden tot progressie en overlijden vergelijkbaar voor patiënten die waren gerandomiseerd naar ARIMIDEX 1 mg en megestrolacetaat. Er zijn in deze gegevens geen aanwijzingen dat ARIMIDEX 10 mg superieur is aan ARIMIDEX 1 mg.
PATIËNT INFORMATIE
ARIMIDEX® A-rim-eh-dex (anastrozol) tabletten voor orale toediening
Wat is de belangrijkste informatie die ik over ARIMIDEX moet weten?
ARIMIDEX kan ernstige bijwerkingen veroorzaken, waaronder:
- hartziekte. Vrouwen met borstkanker in een vroeg stadium, die een voorgeschiedenis hebben van verstopping van hun hartslagaders (ischemische hartziekte) en die ARIMIDEX 1 mg gebruiken, kunnen meer symptomen krijgen van een verminderde bloedtoevoer naar hun hart in vergelijking met vergelijkbare vrouwen die tamoxifen gebruiken.
Roep meteen medische hulp in als u tijdens de behandeling met ARIMIDEX nieuwe of verergerende pijn op de borst of kortademigheid krijgt.
Wat is ARIMIDEX?
ARIMIDEX 1 mg is een receptgeneesmiddel dat wordt gebruikt bij vrouwen na de menopauze (“de verandering van leven”) voor:
- behandeling van vroege borstkanker
- na de operatie
- bij vrouwen bij wie borstkanker hormoonreceptorpositief is
- de eerste behandeling van borstkanker die is uitgezaaid naar nabijgelegen weefsel of lymfeklieren (plaatselijk gevorderd) of is uitgezaaid naar andere delen van het lichaam (gemetastaseerd), bij vrouwen bij wie de borstkanker hormoonreceptorpositief is of de hormoonreceptoren niet bekend zijn
- behandeling van gevorderde borstkanker, als de kanker is gegroeid of de ziekte zich heeft verspreid na behandeling met tamoxifen
ARIMIDEX 1 mg werkt niet bij vrouwen met borstkanker die nog niet in de menopauze zijn geweest (premenopauzale vrouwen).
Wie mag ARIMIDEX niet gebruiken?
Gebruik ARIMIDEX 1 mg niet als u:
- zwanger bent of zwanger kunt worden. ARIMIDEX kan uw ongeboren baby schaden. Als u zwanger wordt terwijl u ARIMIDEX gebruikt, vertel dit dan onmiddellijk aan uw arts.
- niet door de menopauze zijn gegaan (premenopauzaal zijn)
- een ernstige allergische reactie heeft gehad op anastrozol of op een van de bestanddelen van ARIMIDEX. Zie het einde van deze bijsluiter voor een volledige lijst van ingrediënten in ARIMIDEX. Symptomen van een ernstige allergische reactie op ARIMIDEX zijn onder meer: zwelling van het gezicht, de lippen, tong of keel, moeite met ademhalen of slikken, netelroos en jeuk.
Wat moet ik mijn arts vertellen voordat ik ARIMIDEX 1 mg inneem?
Vertel uw arts voordat u ARIMIDEX inneemt als u:
- niet door de menopauze zijn gegaan. Praat met uw arts als u het niet zeker weet.
- een hartprobleem heeft of heeft gehad
- u heeft verteld dat u dunner of zwak bot heeft (osteoporose)
- een hoog cholesterolgehalte hebben
- andere medische aandoeningen heeft
- zwanger bent of van plan bent zwanger te worden. ARIMIDEX kan uw ongeboren baby schaden. Zie "Wie mag ARIMIDEX niet gebruiken?"
- borstvoeding geeft of van plan bent borstvoeding te geven. Het is niet bekend of ARIMIDEX in de moedermelk terechtkomt. U moet samen met uw arts beslissen of u ARIMIDEX 1 mg inneemt of borstvoeding geeft. Je moet niet allebei doen.
Vertel uw arts over alle medicijnen die u gebruikt, inclusief recept- en vrij verkrijgbare medicijnen, vitamines en kruidensupplementen. Vertel het uw arts vooral als u:
- tamoxifen. Je moet niet nemen ARIMIDEX als u tamoxifen gebruikt. Het gebruik van ARIMIDEX 1 mg met tamoxifen kan de hoeveelheid ARIMIDEX 1 mg in uw bloed verlagen en kan ertoe leiden dat ARIMIDEX 1 mg niet zo goed werkt.
- Geneesmiddelen die oestrogeen bevatten. ARIMIDEX 1 mg werkt mogelijk niet als het samen met een van deze geneesmiddelen wordt ingenomen:
- hormoonvervangende therapie
- anticonceptiepillen
- oestrogeen crèmes
- vaginale ringen
- vaginale zetpillen
Weet welke medicijnen u gebruikt. Houd er een lijst van bij om uw arts en apotheker te laten zien wanneer u een nieuw geneesmiddel krijgt.
Hoe moet ik ARIMIDEX innemen?
- Neem ARIMIDEX precies in zoals uw arts u zegt dat u het moet innemen.
- Ga door met het innemen van ARIMIDEX totdat uw arts u zegt te stoppen.
- ARIMIDEX 1 mg kan met of zonder voedsel worden ingenomen.
- Als u een dosis bent vergeten, neem deze dan in zodra u eraan denkt. Als het bijna tijd is voor uw volgende dosis, sla dan de gemiste dosis over. Neem uw volgende regelmatig geplande dosis. Neem geen twee doses tegelijk.
Als u te veel ARIMIDEX 1 mg heeft ingenomen, neem dan onmiddellijk contact op met uw arts of ga naar de eerste hulpafdeling van het dichtstbijzijnde ziekenhuis.
Wat zijn de mogelijke bijwerkingen van ARIMIDEX?
ARIMIDEX kan ernstige bijwerkingen veroorzaken, waaronder:
- Zien “Wat is de belangrijkste informatie die ik over ARIMIDEX moet weten?”
- botverdunning of zwakte (osteoporose). ARIMIDEX 1 mg verlaagt het oestrogeen in uw lichaam, waardoor uw botten dunner en zwakker kunnen worden. Dit kan het risico op fracturen verhogen, vooral van uw wervelkolom, heup en pols. Uw arts kan een botmineraaldichtheidstest bestellen
- voordat u begint en tijdens de behandeling met ARIMIDEX om u te controleren op botveranderingen.
- verhoogd cholesterolgehalte in het bloed (vet in het bloed). Uw arts kan bloedonderzoeken doen om uw cholesterol te controleren terwijl u ARIMIDEX gebruikt.
- huidreacties. Stop met het innemen van ARIMIDEX 1 mg en bel onmiddellijk uw arts als u huidletsels, zweren of blaren krijgt.
- ernstige allergische reacties. Zoek onmiddellijk medische hulp als u:
- zwelling van uw gezicht, lippen, tong of keel
- moeite met slikken of ademen
- lever problemen. ARIMIDEX 1 mg kan een ontsteking van uw lever en veranderingen in het bloedonderzoek van de leverfunctie veroorzaken. Uw arts kan u hierop controleren. Stop met het innemen van ARIMIDEX 1 mg en bel onmiddellijk uw arts als u een van deze tekenen of symptomen van een leverprobleem heeft:
- een algemeen gevoel van niet goed zijn
- geel worden van uw huid of het wit van uw ogen
- pijn aan de rechterkant van uw maagstreek (buik)
Vaak voorkomende bijwerkingen bij vrouwen die ARIMIDEX 1 mg gebruiken, zijn onder meer:
- opvliegers
- zwakheid
- gewrichtspijnen
- gewrichtspijn, stijfheid of zwelling (artritis)
- pijn
- keelpijn
- hoge bloeddruk
- depressie
- misselijkheid en overgeven
- uitslag
- rugpijn
- slaapproblemen
- bot pijn
- hoofdpijn
- zwelling van uw benen, enkels of voeten
- verhoogde hoest
- kortademigheid
- ophoping van lymfevocht in de weefsels van uw aangedane arm (lymfoedeem)
ARIMIDEX kan er ook voor zorgen dat u kietelen, tintelingen of gevoelloosheid van uw huid krijgt.
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van ARIMIDEX. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
Hoe moet ik ARIMIDEX 1 mg bewaren?
- Bewaar ARIMIDEX 1 mg bij kamertemperatuur tussen 20 ° C en 25 ° C (68 ° F tot 77 ° F).
Houd ARIMIDEX en alle geneesmiddelen buiten het bereik van kinderen.
Algemene informatie over het veilige en effectieve gebruik van ARIMIDEX.
Geneesmiddelen worden soms voorgeschreven voor andere doeleinden dan vermeld in een patiëntenbijsluiter. Gebruik ARIMIDEX 1 mg niet voor een aandoening waarvoor het niet is voorgeschreven. Geef ARIMIDEX niet aan andere mensen, ook niet als zij dezelfde symptomen hebben als u. Het kan hen schaden.
Als u meer informatie wilt, neem dan contact op met uw arts. U kunt uw apotheker of arts om informatie vragen over ARIMIDEX die is geschreven voor gezondheidswerkers. Bel voor meer informatie 1-866-992-9276 of ga naar www.ARIMIDEX.com.
Wat zijn de ingrediënten in ARIMIDEX?
Actief ingrediënt: anastrozol
Inactieve ingredienten: lactose, magnesiumstearaat, hydroxypropylmethylcellulose, polyethyleenglycol, povidon, natriumzetmeelglycolaat en titaniumdioxide.