Rebetol 200mg Ribavirin Gebruik, bijwerkingen en dosering. Prijs in online apotheek. Generieke medicijnen zonder recept.
Wat is Rebetol 200 mg en hoe wordt het gebruikt?
Rebetol is een receptgeneesmiddel dat wordt gebruikt om de symptomen van chronische hepatitis C te behandelen. Rebetol 200 mg kan alleen of in combinatie met andere medicijnen worden gebruikt.
Rebetol behoort tot een klasse geneesmiddelen die Hepatitis B/Hepatitis C-agentia worden genoemd; RSV-agenten.
Het is niet bekend of Rebetol 200 mg veilig en effectief is bij kinderen jonger dan 3 jaar.
Wat zijn de mogelijke bijwerkingen van Rebetol?
Rebetol kan ernstige bijwerkingen veroorzaken, waaronder:
- netelroos,
- moeite met ademhalen,
- zwelling van uw gezicht, lippen, tong of keel,
- zichtproblemen
- hevige pijn in uw bovenbuik die zich uitbreidt naar uw rug,
- misselijkheid,
- braken,
- diarree,
- nieuwe of verergerende hoest,
- koorts,
- stekende pijn op de borst,
- piepende ademhaling,
- kortademigheid,
- ernstige depressie,
- gedachten aan zelfbeschadiging,
- gedachten om anderen pijn te doen,
- bleke of vergeelde huid,
- donker gekleurde urine,
- verwardheid,
- zwakheid,
- rillingen,
- griepachtige symptomen,
- gezwollen tandvlees,
- zweertjes in de mond,
- huidzweren,
- gemakkelijk blauwe plekken,
- ongewone bloedingen, en
- duizeligheid
- Roep meteen medische hulp in als u een van de bovenstaande symptomen heeft.
- De meest voorkomende bijwerkingen van Rebetol zijn:
- misselijkheid,
- braken,
- verlies van eetlust,
- koorts,
- rillingen,
- schudden,
- laag aantal bloedcellen,
- Bloedarmoede,
- zwakheid,
- vermoeidheid,
- hoofdpijn,
- spierpijn,
- stemmingswisselingen,
- angst, en
- prikkelbaarheid
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van Rebetol. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
WAARSCHUWING
RISICO OP ERNSTIGE AANDOENINGEN EN RIBAVIRINE-GEASSOCIEERDE EFFECTEN
- REBETOL-monotherapie is niet effectief voor de behandeling van chronische hepatitis C-virusinfectie en mag niet alleen voor deze indicatie worden gebruikt (zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ).
- De primaire toxiciteit van ribavirine is hemolytische anemie. De anemie die gepaard gaat met de behandeling met REBETOL 200 mg kan leiden tot verergering van hartaandoeningen die hebben geleid tot fatale en niet-fatale myocardinfarcten. Patiënten met een voorgeschiedenis van significante of onstabiele hartziekte mogen niet worden behandeld met REBETOL (zie DOSERING EN TOEDIENING, WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en BIJWERKINGEN ).
- Significante teratogene en embryocide effecten zijn aangetoond bij alle diersoorten die aan ribavirine zijn blootgesteld. Bovendien heeft ribavirine een halfwaardetijd van meervoudige doses van 12 dagen en kan het dus tot 6 maanden aanhouden in niet-plasmacompartimenten. Daarom is behandeling met REBETOL gecontra-indiceerd bij vrouwen die zwanger zijn en bij mannelijke partners van vrouwen die zwanger zijn. Bij zowel vrouwelijke patiënten als vrouwelijke partners van mannelijke patiënten die REBETOL-therapie gebruiken, moet uiterste voorzichtigheid worden betracht om zwangerschap te voorkomen tijdens de behandeling en gedurende 6 maanden na voltooiing van de behandeling. Er moeten ten minste twee betrouwbare vormen van effectieve anticonceptie worden gebruikt tijdens de behandeling en tijdens de follow-upperiode van 6 maanden na de behandeling (zie CONTRA-INDICATIES, WAARSCHUWINGEN EN VOORZORGSMAATREGELEN, Gebruik bij specifieke populaties, niet-klinische toxicologie en PATINTINFORMATIE ).
OMSCHRIJVING
REBETOL (ribavirine), is een synthetisch nucleoside-analoog (purine-analoog). De chemische naam van ribavirine is 1-β-D-ribofuranosyl-1H-1,2,4-triazool-3-carboxamide en heeft de volgende structuurformule (zie figuur 1).
Figuur 1: Structuurformule
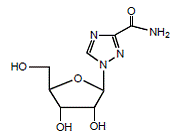
Ribavirine is een wit, kristallijn poeder. Het is goed oplosbaar in water en slecht oplosbaar in watervrije alcohol. De empirische formule is C8H12N4O5 en het molecuulgewicht is 244,21.
REBETOL-capsules bestaan uit een wit poeder in een witte, ondoorzichtige gelatinecapsule. Elke capsule bevat 200 mg ribavirine en de inactieve ingrediënten microkristallijne cellulose, lactosemonohydraat, croscarmellosenatrium en magnesiumstearaat. Het omhulsel van de capsule bestaat uit gelatine, natriumlaurylsulfaat, siliciumdioxide en titaniumdioxide. De capsule is bedrukt met eetbare blauwe farmaceutische inkt die is gemaakt van schellak, watervrije ethylalcohol, isopropylalcohol, n-butylalcohol, propyleenglycol, ammoniumhydroxide en FD&C Blue #2 aluminiumlak.
REBETOL 200 mg drank is een heldere, kleurloze tot bleke of lichtgele vloeistof met kauwgomsmaak. Elke milliliter van de oplossing bevat 40 mg ribavirine en de inactieve ingrediënten sucrose, glycerine, sorbitol, propyleenglycol, natriumcitraat, citroenzuur, natriumbenzoaat, natuurlijke en kunstmatige smaakstof voor kauwgom #15864 en water.
INDICATIES
Chronische hepatitis C (CHC)
REBETOL® (ribavirine) in combinatie met interferon-alfa-2b (gepegyleerd en niet-gepegyleerd) is geïndiceerd voor de behandeling van chronische hepatitis C (CHC) bij patiënten van 3 jaar en ouder met gecompenseerde leverziekte [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , en Gebruik in specifieke populaties ].
De volgende punten moeten in overweging worden genomen bij het starten van REBETOL-combinatietherapie met PegIntron® of INTRON A®:
- Deze indicaties zijn gebaseerd op het bereiken van niet-detecteerbaar HCV-RNA na behandeling gedurende 24 of 48 weken en het behouden van een aanhoudende virologische respons (SVR) 24 weken na de laatste dosis.
- Combinatietherapie met REBETOL/PegIntron heeft de voorkeur boven REBETOL/INTRON A, aangezien deze combinatie aanzienlijk betere responspercentages geeft [zie Klinische studies ].
- Patiënten met de volgende kenmerken hebben minder kans op herbehandeling na het falen van een therapiekuur: eerdere non-respons, eerdere behandeling met gepegyleerd interferon, significante overbruggende fibrose of cirrose en genotype 1-infectie [zie Klinische studies ].
- Er zijn geen veiligheids- en werkzaamheidsgegevens beschikbaar voor een behandeling van langer dan een jaar.
DOSERING EN ADMINISTRATIE
REBETOL-capsules mogen in geen geval worden geopend, geplet of gebroken. REBETOL 200 mg moet met voedsel worden ingenomen [zie KLINISCHE FARMACOLOGIE ]. REBETOL mag niet worden gebruikt bij patiënten met een creatinineklaring van minder dan 50 ml/min.
REBETOL/PegIntron-combinatietherapie
Volwassen patiënten
De aanbevolen dosis PegIntron is 1,5 mcg/kg/week subcutaan in combinatie met 800 tot 1400 mg REBETOL 200 mg capsules oraal op basis van het lichaamsgewicht van de patiënt (zie tabel 1). Het te injecteren volume PegIntron hangt af van de sterkte van PegIntron en het lichaamsgewicht van de patiënt. Raadpleeg de etikettering van PegIntron voor aanvullende doseringsinformatie.
Duur van de behandeling – Interferon-alfa-naïeve patiënten
De behandelingsduur voor patiënten met genotype 1 is 48 weken. Stopzetting van de behandeling dient te worden overwogen bij patiënten die na 12 weken geen daling van ten minste 2 log10 of verlies van HCV-RNA bereiken, of als HCV-RNA na 24 weken therapie nog steeds detecteerbaar is. Patiënten met genotype 2 en 3 moeten gedurende 24 weken worden behandeld.
Duur van de behandeling – Herbehandeling met PegIntron/REBETOL van eerdere mislukte behandelingen
De behandelingsduur voor patiënten bij wie de therapie eerder faalde, is 48 weken, ongeacht het HCV-genotype. Bij herbehandelde patiënten bij wie in week 12 van de therapie geen ondetecteerbaar HCV-RNA wordt bereikt, of bij wie het HCV-RNA na 24 weken therapie nog steeds detecteerbaar is, is het zeer onwaarschijnlijk dat ze SVR bereiken en moet het staken van de behandeling worden overwogen [zie Klinische studies ].
Pediatrische patiënten
De dosering voor pediatrische patiënten wordt bepaald door het lichaamsoppervlak voor PegIntron en door het lichaamsgewicht voor REBETOL. De aanbevolen dosis PegIntron is 60 mcg/m²/week subcutaan in combinatie met 15 mg/kg/dag REBETOL 200 mg oraal in twee verdeelde doses (zie tabel 2) voor pediatrische patiënten in de leeftijd van 3-17 jaar. Patiënten die hun 18e verjaardag bereiken terwijl ze PegIntron/REBETOL 200 mg krijgen, dienen het pediatrische doseringsschema te blijven volgen. De behandelingsduur voor patiënten met genotype 1 is 48 weken. Patiënten met genotype 2 en 3 moeten gedurende 24 weken worden behandeld.
REBETOL/INTRON Een combinatietherapie
volwassenen
Duur van de behandeling – Interferon-alfa-naïeve patiënten
De aanbevolen dosis INTRON A is 3 miljoen IE driemaal per week subcutaan. De aanbevolen dosis REBETOL-capsules hangt af van het lichaamsgewicht van de patiënt (zie tabel 3). De aanbevolen behandelingsduur voor patiënten die niet eerder met interferon zijn behandeld, is 24 tot 48 weken. De duur van de behandeling moet worden aangepast aan de patiënt, afhankelijk van de ziektekenmerken bij aanvang, de respons op de therapie en de verdraagbaarheid van het regime [zie AANWIJZINGEN EN GEBRUIK , ONGEWENSTE REACTIES , en Klinische studies ]. Na 24 weken behandeling moet de virologische respons worden beoordeeld. Stopzetting van de behandeling moet worden overwogen bij elke patiënt die binnen 24 weken geen HCV-RNA heeft bereikt onder de detectielimiet van de test. Er zijn geen veiligheids- en werkzaamheidsgegevens over een behandeling langer dan 48 weken bij de eerder onbehandelde patiëntenpopulatie.
Duur van de behandeling – Herbehandeling met INTRON A/REBETOL bij recidiverende patiënten
Bij patiënten die een terugval krijgen na monotherapie met niet-gepegyleerd interferon, is de aanbevolen behandelingsduur 24 weken.
Kindergeneeskunde
De aanbevolen dosis REBETOL is 15 mg/kg per dag oraal (verdeelde dosis AM en PM). Raadpleeg tabel 2 voor pediatrische dosering van REBETOL in combinatie met INTRON A. INTRON A voor injectie per lichaamsgewicht van 25 kg tot 61 kg is 3 miljoen IE/m² driemaal per week subcutaan. Raadpleeg de doseringstabel voor volwassenen voor een lichaamsgewicht van meer dan 61 kg.
De aanbevolen behandelingsduur is 48 weken voor pediatrische patiënten met genotype 1. Na 24 weken behandeling moet de virologische respons worden beoordeeld. Stopzetting van de behandeling moet worden overwogen bij elke patiënt die tegen die tijd geen HCV-RNA heeft bereikt onder de detectielimiet van de test. De aanbevolen behandelingsduur voor pediatrische patiënten met genotype 2/3 is 24 weken.
Laboratorium testen
De volgende laboratoriumtests worden aanbevolen voor alle patiënten die met REBETOL 200 mg worden behandeld, voorafgaand aan het begin van de behandeling en daarna periodiek.
Standaard hematologische tests - inclusief hemoglobine (voorbehandeling, week 2 en week 4 van de therapie, en indien klinisch aangewezen [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ], het volledige en differentiële aantal witte bloedcellen en het aantal bloedplaatjes.
- Bloedchemie - leverfunctietesten en TSH.
- Zwangerschap - inclusief maandelijkse controle voor vrouwen in de vruchtbare leeftijd.
- ECG [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Dosisaanpassingen
Als zich ernstige bijwerkingen of laboratoriumafwijkingen voordoen tijdens de combinatie van REBETOL/INTRON A-therapie of REBETOL/PegIntron-therapie, wijzig of stop dan de dosis totdat de bijwerking afneemt of in ernst afneemt (zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. Als de intolerantie aanhoudt na dosisaanpassing, moet de combinatietherapie worden stopgezet. Dosisverlaging van PegIntron bij volwassen patiënten die een REBETOL/PegIntron-combinatietherapie ondergaan, wordt bereikt in een proces van twee stappen van de oorspronkelijke startdosis van 1,5 mcg/kg/week, naar 1 mcg/kg/week en vervolgens naar 0,5 mcg/kg/week , indien nodig. Raadpleeg de etikettering van PegIntron voor aanvullende informatie over dosisverlaging van PegIntron.
In de combinatietherapie voor volwassenen, onderzoek 2, traden dosisverlagingen op bij 42% van de proefpersonen die PegIntron 1,5 mcg/kg en REBETOL 800 mg per dag kregen, waaronder 57% van de proefpersonen die 60 kg of minder wogen. In onderzoek 4 had 16% van de proefpersonen een dosisverlaging van PegIntron tot 1 mcg/kg in combinatie met REBETOL, met nog eens 4% die de tweede dosisverlaging van PegIntron tot 0,5 mcg/kg nodig had vanwege bijwerkingen [zie ONGEWENSTE REACTIES ].
Dosisverlaging bij pediatrische patiënten wordt bereikt door de aanbevolen dosis PegIntron in een proces van twee stappen te wijzigen van de oorspronkelijke startdosis van 60 mcg/m²/week naar 40 mcg/m²/week en vervolgens naar 20 mcg/m²/week, indien nodig (zie tabel 4). In de pediatrische combinatietherapiestudie traden dosisverlagingen op bij 25% van de proefpersonen die wekelijks PegIntron 60 mcg/m² en REBETOL 15 mg/kg dagelijks kregen. Dosisverlaging bij pediatrische patiënten wordt bereikt door de aanbevolen dosis REBETOL te wijzigen van de oorspronkelijke startdosis van 15 mg/kg per dag in een tweestaps proces naar 12 mg/kg/dag en vervolgens naar 8 mg/kg/dag, indien nodig ( zie Tabel 4).
REBETOL 200 mg mag niet worden gebruikt bij patiënten met een creatinineklaring van minder dan 50 ml/min. Patiënten met een verminderde nierfunctie en patiënten ouder dan 50 moeten zorgvuldig worden gecontroleerd met betrekking tot de ontwikkeling van bloedarmoede [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , Gebruik bij specifieke populaties , en KLINISCHE FARMACOLOGIE ].
REBETOL 200 mg moet met voorzichtigheid worden toegediend aan patiënten met een reeds bestaande hartziekte. Patiënten moeten vóór aanvang van de therapie worden beoordeeld en moeten tijdens de therapie op passende wijze worden gecontroleerd. Als er enige verslechtering van de cardiovasculaire status is, moet de therapie worden stopgezet [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Voor patiënten met een voorgeschiedenis van stabiele hart- en vaatziekten is een permanente dosisverlaging vereist als de hemoglobinewaarde daalt met meer dan of gelijk aan 2 g/dl gedurende een periode van 4 weken. Bovendien, voor deze patiënten met een hartgeschiedenis, als de hemoglobine na 4 weken op een verlaagde dosis minder dan 12 g/dl blijft, moet de patiënt de combinatietherapie stopzetten.
Het wordt aanbevolen dat een patiënt bij wie het hemoglobinegehalte lager is dan 10 g/dl, zijn/haar dosis REBETOL laat aanpassen of stopzetten volgens tabel 4 [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Raadpleeg de etikettering van INTRON A of PegIntron voor aanvullende informatie over het verlagen van een dosis INTRON A of PegIntron.
Stopzetting van de dosering
volwassenen
Bij HCV-genotype 1, interferon-alfa-naïeve patiënten die PegIntron in combinatie met ribavirine krijgen, wordt stopzetting van de behandeling aanbevolen als er niet ten minste een daling van 2 log10 of verlies van HCV-RNA is na 12 weken therapie, of als HCV-RNA niveaus blijven detecteerbaar na 24 weken therapie. Ongeacht het genotype is het zeer onwaarschijnlijk dat eerder behandelde patiënten met detecteerbaar HCV-RNA in week 12 of 24 SVR bereiken, en stopzetting van de behandeling moet worden overwogen.
Kindergeneeskunde (3-17 jaar)
Het wordt aanbevolen dat patiënten die de combinatie PegIntron/REBETOL krijgen (met uitzondering van HCV-genotype 2 en 3) na 12 weken de behandeling stopzetten als hun behandeling HCV-RNA in week 12 minder dan 2 log10 daalde in vergelijking met een voorbehandeling of na 24 weken als ze detecteerbare HCV-RNA in behandelingsweek 24.
HOE GELEVERD
Doseringsvormen en sterke punten
REBETOL 200 mg capsules 200 mg
REBETOL 200 mg drank 40 mg per ml
Opslag en behandeling
REBETOL 200 mg capsules zijn witte, ondoorzichtige capsules met REBETOL, 200 mg, en het logo van Schering Corporation gedrukt op de capsuleomhulling; de capsules zijn verpakt in een fles met 56 capsules ( NDC 0085-1351-05), 70 capsules ( NDC 0085-1385-07), en 84 capsules ( NDC 0085-1194-03).
REBETOL 200 mg drank 40 mg per ml is een heldere, kleurloze tot bleke of lichtgele vloeistof met kauwgomsmaak en is verpakt in amberkleurige glazen flessen van 4 oz (100 ml/fles) met kindveilige sluitingen ( NDC 0085-1318-01).
De fles REBETOL-capsules moet worden bewaard bij 25°C (77°F); excursies toegestaan tot 15-30°C (59-86°F) [zie USP-gecontroleerde kamertemperatuur ].
REBETOL 200 mg drank moet worden bewaard tussen 2-8°C (36-46°F) of 25°C (77°F); excursies toegestaan tot 15-30°C (59-86°F) [zie USP-gecontroleerde kamertemperatuur ].
REBETOL 200 mg orale oplossing vervaardigd voor: Merck Sharp & Dohme Corp., een dochteronderneming van Whitehouse Station, NJ 08889, VS. Gefabriceerd door: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Canada REBETOL Capsules vervaardigd door: Merck Sharp & Dohme Corp., een dochteronderneming van Whitehouse Station, NJ 08889, VS. Herzien: december 2014.
BIJWERKINGEN
Klinische onderzoeken met REBETOL in combinatie met PegIntron of INTRON A zijn uitgevoerd bij meer dan 7800 proefpersonen in de leeftijd van 3 tot 76 jaar.
De primaire toxiciteit van ribavirine is hemolytische anemie. Verlagingen van de hemoglobinespiegels traden op binnen de eerste 1 tot 2 weken van orale therapie. Hart- en longreacties geassocieerd met anemie kwamen voor bij ongeveer 10% van de patiënten [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Meer dan 96% van alle proefpersonen in klinische onderzoeken ondervond een of meer bijwerkingen. De meest gemelde bijwerkingen bij volwassen proefpersonen die PegIntron of INTRON A in combinatie met REBETOL 200 mg kregen, waren ontsteking/reactie op de injectieplaats, vermoeidheid/asthenie, hoofdpijn, rillingen, koorts, misselijkheid, spierpijn en angst/emotionele labiliteit/prikkelbaarheid. De meest voorkomende bijwerkingen bij pediatrische proefpersonen van 3 jaar en ouder die REBETOL 200 mg kregen in combinatie met PegIntron of INTRON A waren koorts, hoofdpijn, neutropenie, vermoeidheid, anorexie, erytheem op de injectieplaats en braken.
De rubriek Bijwerkingen verwijst naar de volgende klinische onderzoeken:
- REBETOL/PegIntron Combinatietherapie-onderzoeken:
- Klinisch onderzoek 1 – geëvalueerd PegIntron monotherapie (niet verder beschreven in dit label; zie etiket voor PegIntron voor informatie over dit onderzoek).
- Onderzoek 2 – evalueerde REBETOL 800 mg/dag vaste dosis in combinatie met 1,5 mcg/kg/week PegIntron of met INTRON A.
- Onderzoek 3 – evalueerde PegIntron/op gewicht gebaseerde REBETOL 200 mg in combinatie met PegIntron/platte dosis REBETOL 200 mg regime.
- Onderzoek 4 – vergeleek twee doses PegIntron (1,5 mcg/kg/week en 1 mcg/kg/week) in combinatie met REBETOL en een derde behandelgroep die Pegasys® (180 mcg/week)/Copegus® (1000-1200 mg/dag kreeg) ).
- Onderzoek 5 – evalueerde PegIntron (1,5 mcg/kg/week) in combinatie met op gewicht gebaseerde REBETOL bij proefpersonen die bij eerdere behandeling faalden.
- PegIntron/REBETOL 200 mg combinatietherapie bij pediatrische patiënten
- REBETOL/INTRON A Combinatietherapie-onderzoeken voor volwassenen en kindergeneeskunde
Ernstige bijwerkingen zijn opgetreden bij ongeveer 12% van de proefpersonen in klinische onderzoeken met PegIntron met of zonder REBETOL (zie GEVAARLIJKE WAARSCHUWING: , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. De meest voorkomende ernstige voorvallen die optraden bij proefpersonen die werden behandeld met PegIntron en REBETOL 200 mg waren depressie en zelfmoordgedachten [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ], elk met een frequentie van minder dan 1%. Zelfmoordgedachten of zelfmoordpogingen kwamen vaker voor bij pediatrische patiënten, voornamelijk adolescenten, in vergelijking met volwassen patiënten (2,4% versus 1%) tijdens de behandeling en de follow-up buiten de therapie [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. De meest voorkomende fatale reactie die optrad bij proefpersonen die werden behandeld met PegIntron en REBETOL 200 mg was hartstilstand, zelfmoordgedachten en zelfmoordpoging [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ], die allemaal voorkomen bij minder dan 1% van de proefpersonen.
Omdat klinische onderzoeken onder sterk uiteenlopende omstandigheden worden uitgevoerd, kunnen de bijwerkingen die in de klinische onderzoeken van een geneesmiddel zijn waargenomen niet direct worden vergeleken met de percentages in de klinische onderzoeken van een ander geneesmiddel en komen mogelijk niet overeen met de percentages die in de klinische praktijk worden waargenomen.
Ervaring met klinische proeven – REBETOL/PegIntron-combinatietherapie
Volwassen onderwerpen
Bijwerkingen die in de klinische studie optraden met een incidentie van meer dan 5% worden weergegeven per behandelingsgroep uit de REBETOL/PegIntron-combinatietherapie (onderzoek 2) in tabel 5.
Tabel 6 geeft een samenvatting van de behandelingsgerelateerde bijwerkingen in onderzoek 4 die optraden met een incidentie van meer dan of gelijk aan 10%.
De incidentie van ernstige bijwerkingen was in alle onderzoeken vergelijkbaar. In onderzoek 3 werd een vergelijkbare incidentie van ernstige bijwerkingen gemeld voor de REBETOL-groep op basis van gewicht (12%) en voor het REBETOL-regime met een vlakke dosis. In onderzoek 2 was de incidentie van ernstige bijwerkingen 17% in de PegIntron/REBETOL-groepen vergeleken met 14% in de INTRON A/REBETOL-groep.
In veel maar niet alle gevallen verdwenen de bijwerkingen na dosisverlaging of stopzetting van de behandeling. Sommige proefpersonen ondervonden aanhoudende of nieuwe ernstige bijwerkingen tijdens de follow-upperiode van 6 maanden. In onderzoek 2 bleven veel proefpersonen enkele maanden na stopzetting van de therapie bijwerkingen ervaren. Aan het einde van de follow-upperiode van 6 maanden was de incidentie van aanhoudende bijwerkingen per lichaamsklasse in de groep met PegIntron 1,5/REBETOL 200 mg 33% (psychiatrisch), 20% (musculoskeletaal) en 10% (voor endocriene en voor GI). Bij ongeveer 10 tot 15% van de proefpersonen waren gewichtsverlies, vermoeidheid en hoofdpijn niet verdwenen.
Er zijn 31 sterfgevallen bij proefpersonen die optraden tijdens de behandeling of tijdens de follow-up in deze klinische onderzoeken. In onderzoek 1 was er 1 zelfmoord bij een proefpersoon die PegIntron monotherapie kreeg en 2 sterfgevallen bij proefpersonen die INTRON A monotherapie kregen (1 moord/zelfmoord en 1 plotselinge dood). In onderzoek 2 was er 1 zelfmoord bij een proefpersoon die PegIntron/REBETOL 200 mg combinatietherapie kreeg; en 1 patiënt overleden in de INTRON A/REBETOL 200 mg groep (motorvoertuigongeval). In onderzoek 3 waren er 14 sterfgevallen, waarvan 2 vermoedelijke zelfmoorden en 1 onverklaarbare dood bij een persoon met een relevante medische voorgeschiedenis van depressie. In onderzoek 4 waren er 12 sterfgevallen, waarvan 6 bij proefpersonen die combinatietherapie met PegIntron/REBETOL kregen, 5 in de PegIntron 1,5 mcg/REBETOL-arm (N=1019) en 1 in de PegIntron 1 mcg/REBETOL-arm (N= 1016), en waarvan 6 bij proefpersonen die Pegasys/Copegus kregen (N=1035); er waren 3 zelfmoorden die optraden tijdens de follow-upperiode zonder behandeling bij proefpersonen die PegIntron (1,5 mcg/kg)/REBETOL 200 mg combinatietherapie kregen.
In onderzoeken 1 en 2 stopte 10 tot 14% van de proefpersonen die PegIntron kregen, alleen of in combinatie met REBETOL 200 mg, de therapie, vergeleken met 6% behandeld met INTRON A alleen en 13% behandeld met INTRON A in combinatie met REBETOL. Evenzo stopte in onderzoek 3 15% van de proefpersonen die PegIntron kregen in combinatie met REBETOL op gewichtsbasis en 14% van de proefpersonen die PegIntron en een vlakke dosis REBETOL 200 mg kregen de behandeling vanwege een bijwerking. De meest voorkomende redenen voor stopzetting van de behandeling waren gerelateerd aan bekende interferon-effecten van psychiatrische, systemische (bijv. vermoeidheid, hoofdpijn) of gastro-intestinale bijwerkingen. In onderzoek 4 stopte 13% van de proefpersonen in de PegIntron 1,5 mcg/REBETOL 200 mg-arm, 10% in de PegIntron 1 mcg/REBETOL 200 mg-arm en 13% in de Pegasys 180 mcg/Copegus-arm vanwege bijwerkingen.
In onderzoek 2 traden dosisverlagingen als gevolg van bijwerkingen op bij 42% van de proefpersonen die PegIntron (1,5 mcg/kg)/REBETOL 200 mg kregen en bij 34% van de proefpersonen die INTRON A/REBETOL kregen. Bij de meerderheid van de proefpersonen (57%) die 60 kg of minder wogen en die PegIntron (1,5 mcg/kg)/REBETOL kregen, moest de dosis worden verlaagd. Verlaging van interferon was dosisgerelateerd (PegIntron 1,5 mcg/kg hoger dan PegIntron 0,5 mcg/kg of INTRON A), respectievelijk 40%, 27%, 28%. Dosisverlaging voor REBETOL 200 mg was vergelijkbaar in alle drie de groepen, 33 tot 35%. De meest voorkomende redenen voor dosisaanpassingen waren neutropenie (18%), of anemie (9%) (zie: Laboratoriumwaarden ). Andere veel voorkomende redenen waren depressie, vermoeidheid, misselijkheid en trombocytopenie. In onderzoek 3 kwamen dosisaanpassingen als gevolg van bijwerkingen vaker voor bij op gewicht gebaseerde dosering (WBD) dan bij vaste dosering (respectievelijk 29% en 23%). In onderzoek 4 had 16% van de proefpersonen een dosisverlaging van PegIntron tot 1 mcg/kg in combinatie met REBETOL 200 mg, met nog eens 4% die een tweede dosisverlaging van PegIntron tot 0,5 mcg/kg nodig had vanwege bijwerkingen vergeleken met 15% van de proefpersonen in de Pegasys/Copegus-arm, die een dosisverlaging tot 135 mcg/week nodig hadden met Pegasys, met nog eens 7% in de Pegasys/Copegus-arm die een tweede dosisverlaging tot 90 mcg/week met Pegasys nodig had.
In de combinatieonderzoeken met PegIntron/REBETOL 200 mg waren de meest voorkomende bijwerkingen psychiatrisch, die optraden bij 77% van de proefpersonen in onderzoek 2 en 68% tot 69% van de proefpersonen in onderzoek 3. Deze psychiatrische bijwerkingen waren onder meer depressie, prikkelbaarheid en slapeloosheid, elk gemeld door ongeveer 30% tot 40% van de proefpersonen in alle behandelingsgroepen. Suïcidaal gedrag (gedachten, pogingen en zelfmoord) kwam voor bij 2% van alle proefpersonen tijdens de behandeling of tijdens de follow-up na stopzetting van de behandeling [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. In onderzoek 4 traden psychiatrische bijwerkingen op bij 58% van de proefpersonen in de PegIntron 1,5 mcg/REBETOL 200 mg-arm, 55% van de proefpersonen in de PegIntron 1 mcg/REBETOL 200 mg-arm en 57% van de proefpersonen in de Pegasys 180 mcg/Copegus-arm .
PegIntron veroorzaakte vermoeidheid of hoofdpijn bij ongeveer tweederde van de proefpersonen, met koorts of rillingen bij ongeveer de helft van de proefpersonen. De ernst van sommige van deze systemische symptomen (bijv. koorts en hoofdpijn) had de neiging af te nemen naarmate de behandeling werd voortgezet. In onderzoeken 1 en 2 kwamen ontsteking en reactie op de toedieningsplaats (bijv. blauwe plekken, jeuk en irritatie) ongeveer tweemaal zo vaak voor bij PegIntron-therapieën (bij maximaal 75% van de proefpersonen) als bij INTRON A. Pijn op de injectieplaats was echter zeldzaam (2 tot 3%) in alle groepen. In onderzoek 3 was er een algemene incidentie van 23% tot 24% voor reacties op de injectieplaats of ontsteking.
Proefpersonen die REBETOL/PegIntron kregen als herbehandeling nadat een eerdere interferon-combinatiebehandeling had gefaald, meldden bijwerkingen die vergelijkbaar waren met de bijwerkingen die eerder met deze behandeling waren geassocieerd tijdens klinische onderzoeken met niet eerder behandelde proefpersonen.
Pediatrische onderwerpen
Over het algemeen was het bijwerkingenprofiel bij de pediatrische populatie vergelijkbaar met dat waargenomen bij volwassenen. In het pediatrische onderzoek waren de meest voorkomende bijwerkingen bij alle proefpersonen koorts (80%), hoofdpijn (62%), neutropenie (33%), vermoeidheid (30%), anorexia (29%), erytheem op de injectieplaats (29 %) en braken (27%). De meeste bijwerkingen die in het onderzoek werden gemeld, waren licht of matig van ernst. Ernstige bijwerkingen werden gemeld bij 7% (8/107) van alle proefpersonen en omvatten pijn op de injectieplaats (1%), pijn in extremiteit (1%), hoofdpijn (1%), neutropenie (1%) en pyrexie (4 %). Belangrijke bijwerkingen die optraden bij deze populatie waren nervositeit (7%; 7/107), agressie (3%; 3/107), woede (2%; 2/107) en depressie (1%; 1/107) . Vijf proefpersonen kregen een behandeling met levothyroxine, drie met klinische hypothyreoïdie en twee met asymptomatische TSH-verhogingen. De gewichts- en lengtetoename van pediatrische proefpersonen die werden behandeld met PegIntron plus REBETOL bleven achter bij die voorspeld door normatieve populatiegegevens voor de gehele duur van de behandeling. Ernstig geremde groeisnelheid (minder dan 3e percentiel) werd waargenomen bij 70% van de proefpersonen tijdens de behandeling.
Dosisaanpassingen van PegIntron en/of ribavirine waren nodig bij 25% van de proefpersonen vanwege aan de behandeling gerelateerde bijwerkingen, meestal voor anemie, neutropenie en gewichtsverlies. Twee proefpersonen (2%; 2/107) stopten met de behandeling als gevolg van een bijwerking.
Bijwerkingen die optraden met een incidentie van meer dan of gelijk aan 10% bij de pediatrische proefpersonen worden weergegeven in tabel 7.
Vierennegentig van de 107 proefpersonen namen deel aan een 5 jaar durende follow-upstudie. De langetermijneffecten op de groei waren minder bij de proefpersonen die gedurende 24 weken werden behandeld dan bij degenen die gedurende 48 weken werden behandeld. Vierentwintig procent van de proefpersonen (11/46) die gedurende 24 weken werden behandeld en 40% van de proefpersonen (19/48) die gedurende 48 weken werden behandeld, had een afname in lengte-voor-leeftijd van > 15 percentiel vanaf de voorbehandeling tot het einde van 5 jaar lange termijn follow-up vergeleken met de baseline percentielen van voor de behandeling. Bij elf procent van de proefpersonen (5/46) die gedurende 24 weken werden behandeld en bij 13% van de proefpersonen (6/48) die gedurende 48 weken werden behandeld, werd een afname van > 30 lengte-voor-leeftijd-percentielen waargenomen ten opzichte van de uitgangswaarde vóór de behandeling van de 5 jaar lange termijn follow-up. Hoewel waargenomen in alle leeftijdsgroepen, bleek het hoogste risico op verminderde lengte aan het einde van de langetermijnfollow-up te correleren met het starten van combinatietherapie tijdens de jaren van verwachte piekgroeisnelheid. [Zien WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
Laboratoriumwaarden
Volwassen en pediatrische proefpersonen
Het bijwerkingenprofiel in onderzoek 3, waarin de combinatie PegIntron/op gewicht gebaseerde REBETOL 200 mg werd vergeleken met een regime van PegIntron/vlakke dosis REBETOL 200 mg, liet een verhoogde mate van bloedarmoede zien bij dosering op basis van het gewicht (29% vs. 19% voor op gewicht gebaseerde vs. vlakke dosisregimes, respectievelijk). De meeste gevallen van anemie waren echter mild en reageerden op dosisverlagingen.
Veranderingen in geselecteerde laboratoriumwaarden tijdens behandeling in combinatie met behandeling met REBETOL 200 mg worden hieronder beschreven. Verlaging van hemoglobine, leukocyten, neutrofielen en bloedplaatjes kan een dosisverlaging of permanente stopzetting van de therapie vereisen [zien DOSERING EN ADMINISTRATIE ]. Veranderingen in geselecteerde laboratoriumwaarden tijdens de therapie worden beschreven in Tabel 8. De meeste veranderingen in laboratoriumwaarden in het PegIntron/REBETOL 200 mg-onderzoek met pediatrie waren licht of matig.
Hemoglobine
De hemoglobinespiegels daalden tot minder dan 11 g/dl bij ongeveer 30% van de proefpersonen in onderzoek 2. In onderzoek 3 had 47% van de proefpersonen die WBD REBETOL 200 mg kregen en 33% van de proefpersonen die REBETOL 200 mg in een platte dosis kregen een verlaging van het hemoglobinegehalte van minder dan 11 g /dl. Verlagingen van hemoglobine tot minder dan 9 g/dL kwamen vaker voor bij proefpersonen die WBD kregen vergeleken met een vaste dosering (respectievelijk 4% en 2%). In onderzoek 2 was dosisaanpassing vereist bij 9% en 13% van de proefpersonen in de groepen PegIntron/REBETOL 200 mg en INTRON A/REBETOL 200 mg. In onderzoek 4 hadden proefpersonen die PegIntron (1,5 mcg/kg)/REBETOL kregen, verlagingen van de hemoglobinespiegels tot tussen 8,5 en minder dan 10 g/dl (28%) en tot minder dan 8,5 g/dl (3%), terwijl bij patiënten die Pegasys 180 mcg/Copegus kregen, kwamen deze verlagingen voor bij respectievelijk 26% en 4% van de proefpersonen. De hemoglobinespiegels werden gemiddeld stabiel in week 4-6 van de behandeling. Het typische waargenomen patroon was een afname van de hemoglobinespiegels in behandelingsweek 4, gevolgd door stabilisatie en een plateau, dat aanhield tot het einde van de behandeling. In het PegIntron-onderzoek naar monotherapie waren de hemoglobinedalingen over het algemeen licht en waren dosisaanpassingen zelden nodig [zie DOSERING EN ADMINISTRATIE ].
Neutrofielen
Dalingen in het aantal neutrofielen werden waargenomen bij de meeste volwassen proefpersonen die werden behandeld met combinatietherapie met REBETOL 200 mg in onderzoek 2 (85%) en INTRON A/REBETOL (60%). Ernstige potentieel levensbedreigende neutropenie (minder dan 0,5 x 109/l) trad op bij 2% van de proefpersonen die werden behandeld met INTRON A/REBETOL 200 mg en bij ongeveer 4% van de proefpersonen die in onderzoek 2 met PegIntron/REBETOL 200 mg werden behandeld. PegIntron/REBETOL in onderzoek 2 vereiste aanpassing van de interferondosering. Bij weinig proefpersonen (minder dan 1%) moest de behandeling definitief worden stopgezet. Het aantal neutrofielen keerde 4 weken na stopzetting van de therapie over het algemeen terug naar het niveau van voor de behandeling [zie: DOSERING EN ADMINISTRATIE ].
Bloedplaatjes
Het aantal bloedplaatjes daalde tot minder dan 100.000/mm³ bij ongeveer 20% van de proefpersonen die werden behandeld met PegIntron alleen of met REBETOL 200 mg en bij 6% van de volwassen proefpersonen die werden behandeld met INTRON A/REBETOL. Ernstige dalingen van het aantal bloedplaatjes (minder dan 50.000/mm³) komen voor bij minder dan 4% van de volwassen proefpersonen. Patiënten kunnen stopzetting of dosisaanpassing nodig hebben als gevolg van een daling van het aantal bloedplaatjes [zie: DOSERING EN ADMINISTRATIE ]. In onderzoek 2 had 1% of 3% van de proefpersonen dosisaanpassing van respectievelijk INTRON A of PegIntron nodig. Het aantal bloedplaatjes keerde over het algemeen 4 weken na het stoppen van de therapie terug naar het niveau van voor de behandeling.
Schildklierfunctie
De ontwikkeling van TSH-afwijkingen, met of zonder klinische manifestaties, wordt geassocieerd met interferontherapieën. In onderzoek 2 traden klinisch duidelijke schildklieraandoeningen op bij proefpersonen die werden behandeld met INTRON A of PegIntron (met of zonder REBETOL) met een vergelijkbare incidentie (5% voor hypothyreoïdie en 3% voor hyperthyreoïdie). De proefpersonen ontwikkelden nieuwe TSH-afwijkingen tijdens de behandeling en tijdens de follow-upperiode. Aan het einde van de follow-up periode had 7% van de proefpersonen nog steeds afwijkende TSH-waarden.
Bilirubine en urinezuur
In onderzoek 2 ontwikkelde 10 tot 14% van de proefpersonen hyperbilirubinemie en ontwikkelde 33 tot 38% hyperurikemie in verband met hemolyse. Zes proefpersonen ontwikkelden milde tot matige jicht.
Ervaring met klinische proeven – REBETOL/INTRON Een combinatietherapie
Volwassen onderwerpen
In klinische onderzoeken stopte respectievelijk 19% en 6% van de niet eerder behandelde en recidiverende patiënten met de behandeling vanwege bijwerkingen in de combinatie-armen, vergeleken met 13% en 3% in de interferon-armen. Geselecteerde aan de behandeling gerelateerde bijwerkingen die voorkwamen in de Amerikaanse onderzoeken met een incidentie van meer dan of gelijk aan 5%, worden weergegeven per behandelingsgroep (zie tabel 9). Over het algemeen werden de geselecteerde behandelingsgerelateerde bijwerkingen met een lagere incidentie gemeld in de internationale onderzoeken dan in de Amerikaanse onderzoeken, met uitzondering van asthenie, griepachtige symptomen, nervositeit en jeuk.
Pediatrische onderwerpen
In klinische onderzoeken met 118 pediatrische proefpersonen van 3 tot 16 jaar stopte 6% met de behandeling vanwege bijwerkingen. Bij 30% van de proefpersonen waren dosisaanpassingen nodig, meestal voor anemie en neutropenie. Over het algemeen was het bijwerkingenprofiel bij de pediatrische populatie vergelijkbaar met dat waargenomen bij volwassenen. Aandoeningen op de injectieplaats, koorts, anorexia, braken en emotionele labiliteit kwamen vaker voor bij pediatrische proefpersonen dan bij volwassen proefpersonen. Daarentegen ervoeren pediatrische proefpersonen minder vermoeidheid, dyspepsie, artralgie, slapeloosheid, prikkelbaarheid, verminderde concentratie, dyspneu en pruritus in vergelijking met volwassen proefpersonen. Geselecteerde behandelingsgerelateerde bijwerkingen die optraden met een incidentie van meer dan of gelijk aan 5% bij alle pediatrische proefpersonen die de aanbevolen dosis REBETOL/INTRON A-combinatietherapie kregen, worden weergegeven in Tabel 9.
Tijdens een 48-weekse behandelingskuur was er een afname in de snelheid van lineaire groei (gemiddelde percentiele toewijzingsafname van 7%) en een afname in de snelheid van gewichtstoename (gemiddelde percentiele toewijzingsafname van 9%). Een algemene omkering van deze trends werd waargenomen tijdens de periode van 24 weken na de behandeling. Langetermijngegevens bij een beperkt aantal patiënten suggereren echter dat combinatietherapie een groeiremming kan veroorzaken die bij sommige patiënten resulteert in een verminderde uiteindelijke volwassen lengte [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Laboratoriumwaarden
Veranderingen in geselecteerde hematologische waarden (hemoglobine, witte bloedcellen, neutrofielen en bloedplaatjes) tijdens de therapie worden hieronder beschreven (zie Tabel 10).
Hemoglobine. De hemoglobinedalingen bij proefpersonen die REBETOL-therapie kregen, begonnen in week 1, met stabilisatie in week 4. Bij niet eerder behandelde proefpersonen die 48 weken werden behandeld, was de gemiddelde maximale daling ten opzichte van de uitgangswaarde 3,1 g/dl in de Amerikaanse studie en 2,9 g/dl in de internationale proces. Bij patiënten met een recidief was de gemiddelde maximale afname vanaf baseline 2,8 g/dl in de Amerikaanse studie en 2,6 g/dl in de internationale studie. De hemoglobinewaarden keerden bij de meeste proefpersonen binnen 4 tot 8 weken na stopzetting van de therapie terug naar het niveau van voor de behandeling.
Bilirubine en urinezuur. Verhogingen van zowel bilirubine als urinezuur, geassocieerd met hemolyse, werden waargenomen in klinische onderzoeken. De meeste waren matige biochemische veranderingen en werden ongedaan gemaakt binnen 4 weken na stopzetting van de behandeling. Deze waarneming kwam het vaakst voor bij personen met een eerdere diagnose van het syndroom van Gilbert. Dit is niet in verband gebracht met leverdisfunctie of klinische morbiditeit.
Postmarketing-ervaringen
De volgende bijwerkingen zijn vastgesteld en gemeld tijdens het gebruik van REBETOL na goedkeuring in combinatie met INTRON A of PegIntron. Omdat deze reacties vrijwillig worden gemeld door een populatie van onbekende grootte, is het niet altijd mogelijk om op betrouwbare wijze hun frequentie te schatten of een oorzakelijk verband met blootstelling aan geneesmiddelen vast te stellen.
Bloed- en lymfestelselaandoeningen
Zuivere rode-cel-aplasie, aplastische anemie
Oor- en labyrintaandoeningen
Gehoorstoornis, duizeligheid
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen
Pulmonale hypertensie
Oogaandoeningen
Serieuze netvliesloslating
Endocriene aandoeningen
suikerziekte
DRUG-INTERACTIES
Didanosine
Blootstelling aan didanosine of zijn actieve metaboliet (dideoxyadenosine 5'-trifosfaat) wordt verhoogd wanneer didanosine gelijktijdig wordt toegediend met ribavirine, wat klinische toxiciteit kan veroorzaken of verergeren; daarom is gelijktijdige toediening van REBETOL 200 mg capsules of drank en didanosine gecontra-indiceerd. In klinische onderzoeken zijn meldingen van fataal leverfalen, evenals perifere neuropathie, pancreatitis en symptomatische hyperlactatemie/melkzuuracidose gemeld.
Nucleoside-analogen
Leverdecompensatie (sommige fataal) is opgetreden bij patiënten met cirrotische HIV/HCV-co-infectie die een antiretrovirale combinatietherapie voor HIV en interferon-alfa en ribavirine kregen. Het toevoegen van een behandeling met alfa-interferonen alleen of in combinatie met ribavirine kan het risico bij deze patiëntenpopulatie verhogen. Patiënten die interferon met ribavirine en nucleoside reverse-transcriptaseremmers (NRTI's) krijgen, moeten nauwlettend worden gecontroleerd op met de behandeling samenhangende toxiciteiten, met name leverdecompensatie en anemie. Stopzetting van NRTI's moet als medisch passend worden beschouwd (zie: etikettering voor individueel NRTI-product ). Dosisverlaging of stopzetting van interferon, ribavirine of beide moet ook worden overwogen als verergering van klinische toxiciteiten wordt waargenomen, waaronder leverdecompensatie (bijv. Child-Pugh groter dan 6).
Ribavirine kan de antivirale activiteit van stavudine en zidovudine in celculturen tegen HIV tegenwerken. In celkweken is aangetoond dat ribavirine de fosforylering van lamivudine, stavudine en zidovudine remt, wat kan leiden tot verminderde antiretrovirale activiteit. In een onderzoek met een ander gepegyleerd interferon in combinatie met ribavirine werd echter geen farmacokinetische (bijv. plasmaconcentraties of intracellulaire concentraties van de actieve metabolieten van trifosforyl) of farmacodynamische (bijv. verlies van hiv/HCV-virologische onderdrukking) waargenomen wanneer ribavirine en lamivudine (n = 18), stavudine (n=10) of zidovudine (n=6) werden gelijktijdig toegediend als onderdeel van een multidrugregime bij personen met een gelijktijdige HIV/HCV-infectie. Daarom moet gelijktijdig gebruik van ribavirine met een van deze geneesmiddelen met voorzichtigheid worden gebruikt.
Geneesmiddelen gemetaboliseerd door cytochroom P-450
Resultaten van in-vitro-onderzoeken waarbij zowel menselijke als rattenlevermicrosoompreparaten werden gebruikt, wezen op weinig of geen door cytochroom P-450-enzym gemedieerd metabolisme van ribavirine, met een minimaal potentieel voor op P-450-enzym gebaseerde geneesmiddelinteracties.
Er werden geen farmacokinetische interacties waargenomen tussen INTRON A en REBETOL 200 mg capsules in een farmacokinetisch onderzoek met meerdere doses.
azathioprine
Van het gebruik van ribavirine voor de behandeling van chronische hepatitis C bij patiënten die azathioprine krijgen, is gemeld dat het ernstige pancytopenie veroorzaakt en het risico op azathioprine-gerelateerde myelotoxiciteit kan verhogen. Inosinemonofosfaatdehydrogenase (IMDH) is vereist voor een van de metabole routes van azathioprine. Van ribavirine is bekend dat het IMDH remt, wat leidt tot accumulatie van een azathioprine-metaboliet, 6-methylthioinosinemonofosfaat (6-MTITP), wat in verband wordt gebracht met myelotoxiciteit (neutropenie, trombocytopenie en anemie). Bij patiënten die azathioprine met ribavirine krijgen, moet het volledige bloedbeeld, inclusief het aantal bloedplaatjes, wekelijks worden gecontroleerd gedurende de eerste maand, tweemaal per maand gedurende de tweede en derde maand van de behandeling, daarna maandelijks of vaker als dosering of andere therapiewijzigingen nodig zijn [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
WAARSCHUWINGEN
Inbegrepen als onderdeel van de PREVENTIEVE MAATREGELEN sectie.
PREVENTIEVE MAATREGELEN
Zwangerschap
REBETOL 200 mg capsules en drank kunnen geboorteafwijkingen en de dood van het ongeboren kind veroorzaken. Behandeling met REBETOL mag niet worden gestart voordat een melding van een negatieve zwangerschapstest is verkregen, onmiddellijk voorafgaand aan de geplande start van de therapie. Patiënten dienen ten minste twee vormen van anticonceptie te gebruiken en maandelijkse zwangerschapstests te ondergaan tijdens de behandeling en gedurende de periode van 6 maanden nadat de behandeling is gestopt. Uiterste zorg moet worden betracht om zwangerschap bij vrouwelijke patiënten en bij vrouwelijke partners van mannelijke patiënten te voorkomen. REBETOL 200mg heeft aangetoond significante teratogene en embryocide effecten bij alle diersoorten waarin adequate studies zijn uitgevoerd. Deze effecten traden op bij doses als laag als een twintigste van de aanbevolen dosis ribavirine voor de mens. De behandeling met REBETOL 200 mg mag pas worden gestart na een melding van een negatieve zwangerschapstest is verkregen onmiddellijk voorafgaand aan de geplande start van de therapie [zie: GEVAARLIJKE WAARSCHUWING: , CONTRA-INDICATIES , Gebruik bij specifieke populaties , en PATIËNT INFORMATIE ].
Bloedarmoede
De primaire toxiciteit van ribavirine is hemolytische anemie, die werd waargenomen bij ongeveer 10% van de met REBETOL/INTRON A behandelde proefpersonen in klinische onderzoeken. De anemie die gepaard gaat met REBETOL-capsules treedt op binnen 1 tot 2 weken na aanvang van de therapie. Omdat de initiële daling van het hemoglobine aanzienlijk kan zijn, wordt geadviseerd hemoglobine of hematocriet te verkrijgen vóór het begin van de behandeling en in week 2 en week 4 van de behandeling, of vaker indien klinisch geïndiceerd. Patiënten moeten dan worden gevolgd zoals klinisch aangewezen [zie: DOSERING EN ADMINISTRATIE ].
Fatale en niet-fatale myocardinfarcten zijn gemeld bij patiënten met anemie veroorzaakt door REBETOL. Patiënten moeten worden beoordeeld op onderliggende hartziekte voordat de behandeling met ribavirine wordt gestart. Bij patiënten met een reeds bestaande hartziekte dienen vóór de behandeling elektrocardiogrammen te worden toegediend en dienen deze tijdens de behandeling adequaat te worden gecontroleerd. Als er enige verslechtering van de cardiovasculaire status is, moet de therapie worden gestaakt of stopgezet [zie: DOSERING EN ADMINISTRATIE ]. Omdat hartaandoeningen kunnen verergeren door geneesmiddelgeïnduceerde anemie, mogen patiënten met een voorgeschiedenis van significante of onstabiele hartaandoeningen REBETOL niet gebruiken.
Pancreatitis
Behandeling met REBETOL en INTRON A of PegIntron moet worden onderbroken bij patiënten met tekenen en symptomen van pancreatitis en worden stopgezet bij patiënten met bevestigde pancreatitis.

Longaandoeningen
Pulmonale symptomen, waaronder dyspneu, longinfiltraten, pneumonitis, pulmonale hypertensie en pneumonie, zijn gemeld tijdens behandeling met REBETOL met combinatietherapie met alfa-interferon; incidentele gevallen van fatale pneumonie zijn voorgekomen. Daarnaast is sarcoïdose of de verergering van sarcoïdose gemeld. Als er aanwijzingen zijn voor longinfiltraten of een verminderde longfunctie, moet de patiënt nauwlettend worden gecontroleerd en indien nodig moet de combinatietherapie worden stopgezet.
Oogaandoeningen
Ribavirine wordt gebruikt in combinatietherapie met alfa-interferonen. Vermindering of verlies van gezichtsvermogen, retinopathie inclusief macula-oedeem, retinale slagader of ader, trombose, retinale bloedingen en wattenvlekken, optische neuritis, papiloedeem en sereuze netvliesloslating worden veroorzaakt of verergerd door behandeling met alfa-interferonen. Alle patiënten dienen bij aanvang een oogonderzoek te ondergaan. Patiënten met reeds bestaande oftalmologische aandoeningen (bijv. diabetische of hypertensieve retinopathie) dienen periodiek oogheelkundig onderzoek te ondergaan tijdens combinatietherapie met behandeling met alfa-interferon. Elke patiënt die oogsymptomen ontwikkelt, moet een onmiddellijk en volledig oogonderzoek ondergaan. Combinatietherapie met alfa-interferonen moet worden stopgezet bij patiënten die nieuwe of verergerende oogaandoeningen ontwikkelen.
Laboratorium testen
PegIntron in combinatie met ribavirine kan ernstige verlagingen van het aantal neutrofielen en bloedplaatjes en hematologische, endocriene (bijv. TSH) en leverafwijkingen veroorzaken.
Bij patiënten die een combinatietherapie met PegIntron/REBETOL ondergaan, dienen hematologie- en bloedchemietests te worden uitgevoerd vóór het begin van de behandeling en daarna periodiek. In het klinische onderzoek bij volwassenen werden volledige bloedtellingen (inclusief hemoglobine-, neutrofielen- en bloedplaatjesaantallen) en chemie (inclusief AST, ALT, bilirubine en urinezuur) gemeten tijdens de behandelingsperiode in week 2, 4, 8, 12 en daarna met tussenpozen van 6 weken, of vaker als zich afwijkingen ontwikkelden. Bij pediatrische proefpersonen werden dezelfde laboratoriumparameters geëvalueerd met aanvullende beoordeling van hemoglobine in behandelingsweek 6. TSH-spiegels werden elke 12 weken gemeten tijdens de behandelingsperiode. HCV-RNA moet tijdens de behandeling periodiek worden gemeten [zie: DOSERING EN ADMINISTRATIE ].
Tand- en parodontale aandoeningen
Tand- en parodontale aandoeningen zijn gemeld bij patiënten die een combinatietherapie met ribavirine en interferon of peginterferon kregen. Bovendien kan een droge mond een schadelijk effect hebben op tanden en slijmvliezen van de mond tijdens langdurige behandeling met de combinatie van REBETOL en gepegyleerd of niet-gepegyleerd interferon-alfa-2b. Patiënten moeten hun tanden tweemaal per dag grondig poetsen en regelmatig gebitsonderzoeken ondergaan. Als ze moeten braken, moeten ze het advies krijgen om daarna hun mond grondig te spoelen.
Gelijktijdige toediening van azathioprine
In de literatuur is gemeld dat pancytopenie (aanzienlijke afname van het aantal rode bloedcellen, neutrofielen en bloedplaatjes) en beenmergsuppressie optreden binnen 3 tot 7 weken na gelijktijdige toediening van gepegyleerd interferon/ribavirine en azathioprine. Bij dit beperkte aantal patiënten (n=8) was myelotoxiciteit reversibel binnen 4 tot 6 weken na stopzetting van zowel antivirale HCV-therapie als gelijktijdige azathioprine en trad niet opnieuw op na herstart van een van beide behandelingen alleen. PegIntron, REBETOL en azathioprine moeten worden stopgezet voor pancytopenie en gepegyleerd interferon/ribavirine mag niet opnieuw worden geïntroduceerd met gelijktijdige toediening van azathioprine (zie DRUG-INTERACTIES ].
Impact op groei - pediatrisch gebruik
Gegevens over de effecten van PegIntron en REBETOL op de groei zijn afkomstig van een open-label onderzoek bij proefpersonen van 3 tot en met 17 jaar, waarin veranderingen in gewicht en lengte worden vergeleken met normatieve populatiegegevens in de VS. Over het algemeen blijven de gewichts- en lengtetoename van pediatrische patiënten die met PegIntron en REBETOL 200 mg worden behandeld, achter bij die voorspeld door normatieve populatiegegevens voor de gehele duur van de behandeling. Ernstig geremde groeisnelheid (minder dan 3e percentiel) werd waargenomen bij 70% van de proefpersonen tijdens de behandeling. Na de behandeling traden bij de meeste proefpersonen reboundgroei en gewichtstoename op. Langetermijn-follow-upgegevens bij pediatrische proefpersonen geven echter aan dat PegIntron in combinatietherapie met REBETOL een groeiremming kan veroorzaken die bij sommige patiënten resulteert in verminderde volwassen lengte [zie ONGEWENSTE REACTIES ].
Evenzo werd een effect op de groei waargenomen bij proefpersonen na een jaar behandeling met REBETOL 200 mg en INTRON A combinatietherapie. In een langdurige follow-upstudie met een beperkt aantal van deze proefpersonen resulteerde combinatietherapie bij sommige proefpersonen in een verminderde uiteindelijke volwassen lengte [zie ONGEWENSTE REACTIES ].
Gebruikswaarborgen
Op basis van de resultaten van klinische onderzoeken is monotherapie met ribavirine niet effectief voor de behandeling van chronische hepatitis C-virusinfectie; daarom mag REBETOL 200 mg capsules of drank niet alleen worden gebruikt. De veiligheid en werkzaamheid van REBETOL capsules en drank zijn alleen vastgesteld bij gebruik in combinatie met INTRON A of PegIntron (geen andere interferonen) als combinatietherapie.
De veiligheid en werkzaamheid van REBETOL/INTRON A- en PegIntron-therapie voor de behandeling van HIV-infectie, adenovirus, RSV, para-influenza of influenza-infecties zijn niet vastgesteld. REBETOL 200 mg capsules mogen niet worden gebruikt voor deze indicaties. Ribavirine voor inhalatie heeft een aparte etikettering, die moet worden geraadpleegd als inhalatietherapie met ribavirine wordt overwogen.
Er zijn significante bijwerkingen veroorzaakt door behandeling met REBETOL/INTRON A of PegIntron, waaronder ernstige depressie en zelfmoordgedachten, hemolytische anemie, onderdrukking van de beenmergfunctie, auto-immuun- en infectieziekten, longdisfunctie, pancreatitis en diabetes. Zelfmoordgedachten of zelfmoordpogingen kwamen vaker voor bij pediatrische patiënten, voornamelijk adolescenten, in vergelijking met volwassen patiënten (2,4% versus 1%) tijdens de behandeling en de follow-up buiten de therapie. De etikettering voor INTRON A en PegIntron moet in hun geheel worden herzien voor aanvullende veiligheidsinformatie voordat met de combinatiebehandeling wordt begonnen.
Informatie over patiëntbegeleiding
Adviseer de patiënt om de door de FDA goedgekeurde patiëntetikettering te lezen ( Medicatiegids ).
Bloedarmoede
De meest voorkomende bijwerking die optreedt bij REBETOL-capsules is bloedarmoede, die ernstig kan zijn [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en ONGEWENSTE REACTIES ]. Patiënten moeten erop worden gewezen dat laboratoriumonderzoeken nodig zijn voordat de behandeling wordt gestart en periodiek daarna [zie: DOSERING EN ADMINISTRATIE ]. Het wordt aanbevolen dat patiënten goed gehydrateerd zijn, vooral tijdens de beginfase van de behandeling.
Zwangerschap
Patiënten moeten erop worden gewezen dat REBETOL 200 mg capsules en drank geboorteafwijkingen en de dood van het ongeboren kind kunnen veroorzaken. REBETOL 200 mg mag niet worden gebruikt door vrouwen die zwanger zijn of door mannen van wie de vrouwelijke partners zwanger zijn. Uiterste voorzichtigheid moet worden betracht om zwangerschap te voorkomen bij vrouwelijke patiënten en bij vrouwelijke partners van mannelijke patiënten die REBETOL gebruiken. Er mag niet met REBETOL worden gestart totdat een melding van een negatieve zwangerschapstest is verkregen, onmiddellijk voorafgaand aan de start van de behandeling. Patiënten moeten maandelijks een zwangerschapstest uitvoeren tijdens de therapie en gedurende 6 maanden na de therapie.
Vrouwen die zwanger kunnen worden, moeten worden geadviseerd over het gebruik van effectieve anticonceptie (twee betrouwbare vormen) voordat met de behandeling wordt begonnen. Patiënten (mannen en vrouwen) moeten worden geïnformeerd over de teratogene/embryocidale risico's en moeten worden geïnstrueerd om effectieve anticonceptie toe te passen tijdens REBETOL 200 mg en gedurende 6 maanden na de behandeling. Patiënten (mannen en vrouwen) moeten worden geadviseerd om de arts onmiddellijk op de hoogte te stellen in het geval van een zwangerschap [zie: CONTRA-INDICATIES , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , en Gebruik in specifieke populaties ].
Als zwangerschap optreedt tijdens de behandeling of gedurende 6 maanden na de behandeling, moet de patiënt worden geïnformeerd over het teratogene risico van behandeling met REBETOL 200 mg voor de foetus. Patiënten, of partners van patiënten, moeten elke zwangerschap die optreedt tijdens de behandeling of binnen 6 maanden na het staken van de behandeling onmiddellijk aan hun arts melden. Voorschrijvers moeten dergelijke gevallen melden door te bellen naar 1-800-593-2214.
Risico's versus voordelen
Patiënten die REBETOL-capsules krijgen, moeten worden geïnformeerd over de voordelen en risico's van de behandeling, gericht zijn op het juiste gebruik en doorverwezen worden naar de patiënt Medicatiegids . Patiënten moeten worden geïnformeerd dat het effect van de behandeling van een hepatitis C-infectie op de overdracht niet bekend is en dat passende voorzorgsmaatregelen moeten worden genomen om de overdracht van het hepatitis C-virus te voorkomen.
Patiënten moeten worden geïnformeerd over wat ze moeten doen als ze een dosis REBETOL missen; de gemiste dosis moet zo snel mogelijk op dezelfde dag worden ingenomen. Patiënten mogen de volgende dosis niet verdubbelen. Patiënten moeten worden geadviseerd om contact op te nemen met hun zorgverlener als ze vragen hebben.
Niet-klinische toxicologie
Carcinogenese, mutagenese, verminderde vruchtbaarheid
Carcinogenese
Ribavirine veroorzaakte geen toename van enig tumortype wanneer het gedurende 6 maanden werd toegediend in het transgene p53-deficiënte muismodel in doses tot 300 mg/kg (geschat humaan equivalent van 25 mg/kg op basis van aanpassing van het lichaamsoppervlak voor een volwassene van 60 kg ongeveer 1,9 maal de maximaal aanbevolen dagelijkse dosis voor mensen). Ribavirine was niet-carcinogeen wanneer het gedurende 2 jaar aan ratten werd toegediend in doses tot 40 mg/kg (geschat humaan equivalent van 5,71 mg/kg op basis van aanpassing van het lichaamsoppervlak voor een volwassene van 60 kg).
Mutagenese
Ribavirine vertoonde verhoogde incidenties van mutatie en celtransformatie in meerdere genotoxiciteitstests. Ribavirine was actief in de Balb/3T3 in vitro celtransformatieassay. Mutagene activiteit werd waargenomen in de lymfoomtest bij muizen en bij doses van 20 tot 200 mg/kg (geschat humaan equivalent van 1,67 tot 16,7 mg/kg, gebaseerd op aanpassing van het lichaamsoppervlak voor een volwassene van 60 kg; 0,1 tot 1 keer het maximum aanbevolen humane 24-uurs dosis ribavirine) in een micronucleustest bij muizen. Een dominante letale test bij ratten was negatief, wat aangeeft dat als mutaties bij ratten optraden, deze niet werden overgedragen via mannelijke gameten.
Aantasting van de vruchtbaarheid
Ribavirine vertoonde significante embryocidale en teratogene effecten bij doses die ver onder de aanbevolen dosis voor de mens liggen bij alle diersoorten waarin adequate onderzoeken zijn uitgevoerd. Misvormingen van de schedel, het gehemelte, het oog, de kaak, de ledematen, het skelet en het maagdarmkanaal werden opgemerkt. De incidentie en ernst van teratogene effecten namen toe naarmate de dosis van het geneesmiddel werd verhoogd. De overleving van foetussen en nakomelingen was verminderd. In conventionele embryotoxiciteits-/teratogeniteitsstudies bij ratten en konijnen waren de dosisniveaus zonder effect duidelijk lager dan die voor voorgesteld klinisch gebruik (0,3 mg/kg/dag voor zowel ratten als konijnen; ongeveer 0,06 maal de aanbevolen 24-uursdosis van ribavirine). Er werden geen maternale toxiciteit of effecten op het nageslacht waargenomen in een peri-/postnatale toxiciteitsstudie bij ratten die oraal werden gedoseerd tot 1 mg/kg/dag (geschatte humane equivalente dosis van 0,17 mg/kg gebaseerd op aanpassing van het lichaamsoppervlak voor een volwassene van 60 kg ongeveer 0,01 maal de maximaal aanbevolen 24-uurs dosis ribavirine voor mensen (zie: CONTRA-INDICATIES , en WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Vruchtbare vrouwen en partners van vruchtbare vrouwen mogen geen REBETOL krijgen tenzij de patiënt en zijn/haar partner effectieve anticonceptie gebruiken (twee betrouwbare vormen). Op basis van een meervoudige halfwaardetijd (t1/2) van ribavirine van 12 dagen, moet effectieve anticonceptie worden gebruikt gedurende 6 maanden na de therapie (bijv. 15 klaringshalfwaardetijden voor ribavirine).
REBETOL moet met voorzichtigheid worden gebruikt bij vruchtbare mannen. In studies bij muizen om het tijdsverloop en de reversibiliteit van door ribavirine geïnduceerde testiculaire degeneratie te evalueren bij doses van 15 tot 150 mg/kg/dag (geschat humaan equivalent van 1,25 tot 12,5 mg/kg/dag, gebaseerd op aanpassing van het lichaamsoppervlak voor een volwassene van 60 kg; 0,1-0,8 maal de maximale humane 24-uurs dosis ribavirine) toegediend gedurende 3 of 6 maanden, kwamen er afwijkingen in het sperma voor. Na stopzetting van de behandeling was vrijwel volledig herstel van door ribavirine geïnduceerde testiculaire toxiciteit zichtbaar binnen 1 of 2 spermatogenesecycli.
Gebruik bij specifieke populaties
Zwangerschap
Zwangerschap Categorie X
[Zien CONTRA-INDICATIES , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , en Niet-klinische toxicologie ].
Behandeling en nabehandeling:
Potentieel risico voor de foetus
Van ribavirine is bekend dat het zich ophoopt in intracellulaire componenten van waaruit het zeer langzaam wordt geklaard. Het is niet bekend of ribavirine in sperma een mogelijk teratogeen effect zal hebben bij bevruchting van de eicellen. In een onderzoek bij ratten werd geconcludeerd dat dominante letaliteit niet werd geïnduceerd door ribavirine in doses tot 200 mg/kg gedurende 5 dagen (geschatte humane equivalente doses van 7,14 tot 28,6 mg/kg, gebaseerd op aanpassing van het lichaamsoppervlak voor een 60 kg volwassene; tot 1,7 maal de maximaal aanbevolen dosis ribavirine voor de mens). Vanwege de mogelijke teratogene effecten van ribavirine bij de mens, moeten mannelijke patiënten echter worden geadviseerd alle voorzorgsmaatregelen te nemen om het risico op zwangerschap voor hun vrouwelijke partners te voorkomen.
Vrouwen die zwanger kunnen worden, mogen geen REBETOL krijgen tenzij ze tijdens de behandelingsperiode effectieve anticonceptie gebruiken (twee betrouwbare vormen). Bovendien dient effectieve anticonceptie te worden gebruikt gedurende 6 maanden na de therapie op basis van een halfwaardetijd (t1/2) van ribavirine bij meervoudige doses van 12 dagen.
Mannelijke patiënten en hun vrouwelijke partners moeten effectieve anticonceptie toepassen (twee betrouwbare vormen) tijdens de behandeling met REBETOL en gedurende de periode van 6 maanden na de therapie (bijv. 15 halfwaardetijden voor de klaring van ribavirine uit het lichaam).
Er is een Ribavirine-zwangerschapsregister opgesteld om de maternale-foetale uitkomsten van zwangerschappen bij vrouwelijke patiënten en vrouwelijke partners van mannelijke patiënten die zijn blootgesteld aan ribavirine tijdens de behandeling en gedurende 6 maanden na beëindiging van de behandeling, te controleren. Artsen en patiënten worden aangemoedigd om dergelijke gevallen te melden door te bellen naar 1-800-593-2214.
Moeders die borstvoeding geven
Het is niet bekend of het REBETOL-product wordt uitgescheiden in de moedermelk. Vanwege de mogelijkheid van ernstige bijwerkingen van het geneesmiddel bij zuigelingen die borstvoeding geven, moet worden besloten of de borstvoeding moet worden gestaakt of dat REBETOL moet worden uitgesteld of gestaakt.
Pediatrisch gebruik
De veiligheid en werkzaamheid van REBETOL 200 mg in combinatie met PegIntron is niet vastgesteld bij pediatrische patiënten jonger dan 3 jaar. Voor behandeling met REBETOL/INTRON A moet rekening worden gehouden met bewijs van ziekteprogressie, zoals leverontsteking en fibrose, evenals prognostische factoren voor respons, HCV-genotype en virale belasting bij de beslissing om een pediatrische patiënt te behandelen. De voordelen van de behandeling moeten worden afgewogen tegen de waargenomen veiligheidsbevindingen.
Opvolggegevens op lange termijn bij pediatrische proefpersonen geven aan dat REBETOL in combinatie met PegIntron of met INTRON A een groeiremming kan veroorzaken die bij sommige patiënten resulteert in verminderde lengte [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en ONGEWENSTE REACTIES ].
Zelfmoordgedachten of zelfmoordpogingen kwamen vaker voor bij pediatrische patiënten, voornamelijk adolescenten, in vergelijking met volwassen patiënten (2,4% vs. 1%) tijdens de behandeling en de follow-up buiten de therapie [zien WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. Net als bij volwassen patiënten ondervonden pediatrische patiënten andere psychiatrische bijwerkingen (bijv. depressie, emotionele labiliteit, slaperigheid), bloedarmoede en neutropenie [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Geriatrisch gebruik
Klinische onderzoeken naar de behandeling met REBETOL/INTRON A of PegIntron omvatten niet voldoende aantallen proefpersonen van 65 jaar en ouder om te bepalen of zij anders reageren dan jongere proefpersonen.
Van REBETOL 200 mg is bekend dat het grotendeels door de nieren wordt uitgescheiden en het risico op toxische reacties op dit geneesmiddel kan groter zijn bij patiënten met een verminderde nierfunctie. Omdat oudere patiënten vaak een verminderde nierfunctie hebben, moet voorzichtigheid worden betracht bij het kiezen van de dosering. De nierfunctie moet worden gecontroleerd en de dosering moet dienovereenkomstig worden aangepast. REBETOL 200 mg mag niet worden gebruikt bij patiënten met een creatinineklaring van minder dan 50 ml/min [zie: CONTRA-INDICATIES ].
Over het algemeen dienen REBETOL 200 mg capsules met voorzichtigheid te worden toegediend aan oudere patiënten, beginnend aan de onderkant van het doseringsbereik, als gevolg van de grotere frequentie van verminderde lever- en hartfunctie en van gelijktijdige ziekte of andere medicamenteuze behandeling. In klinische onderzoeken hadden oudere proefpersonen een hogere frequentie van bloedarmoede (67%) dan jongere patiënten (28%) [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Ontvangers van orgaantransplantaties
De veiligheid en werkzaamheid van INTRON A en PegIntron alleen of in combinatie met REBETOL voor de behandeling van hepatitis C bij ontvangers van lever- of andere orgaantransplantaties zijn niet vastgesteld. In een kleine (n=16) single-center, ongecontroleerde casus, kwam nierfalen bij ontvangers van niertransplantaten die een combinatietherapie met interferon alfa en ribavirine kregen vaker voor dan verwacht op grond van de eerdere ervaring van het centrum met ontvangers van niertransplantaten die geen combinatietherapie kregen. De relatie tussen nierfalen en afstoting van niertransplantaten is niet duidelijk.
HIV- of HBV-co-infectie
De veiligheid en werkzaamheid van PegIntron/REBETOL 200 mg en INTRON A/REBETOL voor de behandeling van patiënten met HCV die tegelijkertijd met HIV of HBV zijn geïnfecteerd, zijn niet vastgesteld.
OVERDOSERING
Er is beperkte ervaring met overdosering. Acute inname van maximaal 20 g REBETOL-capsules, INTRON A-opname van maximaal 120 miljoen eenheden en subcutane doses INTRON A tot 10 keer de aanbevolen doses zijn gemeld. De primaire effecten die zijn waargenomen, zijn een verhoogde incidentie en ernst van de bijwerkingen die verband houden met het therapeutische gebruik van INTRON A en REBETOL. Leverenzymafwijkingen, nierfalen, bloeding en myocardinfarct zijn echter gemeld bij toediening van enkelvoudige subcutane doses INTRON A die de doseringsaanbevelingen overschrijden.
Er is geen specifiek antidotum voor een overdosis INTRON A of REBETOL 200 mg, en hemodialyse en peritoneale dialyse zijn niet effectief voor de behandeling van een overdosis van deze middelen.
CONTRA-INDICATIES
REBETOL combinatietherapie is gecontra-indiceerd bij:
- vrouwen die zwanger zijn. REBETOL 200 mg kan schade aan de foetus veroorzaken wanneer het wordt toegediend aan een zwangere vrouw. REBETOL is gecontra-indiceerd bij vrouwen die zwanger zijn of kunnen worden. Als REBETOL tijdens de zwangerschap wordt gebruikt, of als de patiënte zwanger wordt terwijl ze REBETOL 200 mg gebruikt, moet de patiënte worden geïnformeerd over het mogelijke gevaar voor haar foetus [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , Gebruik in specifieke populaties , en PATIËNT INFORMATIE ]
- mannen van wie de vrouwelijke partners zwanger zijn
- patiënten met bekende overgevoeligheidsreacties zoals Stevens-Johnson-syndroom, toxische epidermale necrolyse en erythema multiforme op ribavirine of een van de bestanddelen van het product
- patiënten met auto-immuunhepatitis
- patiënten met hemoglobinopathieën (bijv. thalassemie major, sikkelcelanemie)
- patiënten met een creatinineklaring van minder dan 50 ml/min. [zien Gebruik in specifieke populaties en KLINISCHE FARMACOLOGIE ]
- Gelijktijdige toediening van REBETOL en didanosine is gecontra-indiceerd omdat de blootstelling aan de actieve metaboliet van didanosine (dideoxyadenosine 5'-trifosfaat) verhoogd is. Fataal leverfalen, evenals perifere neuropathie, pancreatitis en symptomatische hyperlactatemie/lactaatacidose zijn gemeld bij patiënten die didanosine in combinatie met ribavirine kregen (zie DRUG-INTERACTIES ].
KLINISCHE FARMACOLOGIE
Werkingsmechanisme
Ribavirine is een antiviraal middel [zie Microbiologie ].
Farmacokinetiek
De farmacokinetische eigenschappen van enkelvoudige en meervoudige doses bij volwassenen zijn samengevat in Tabel 11. Ribavirine werd snel en uitgebreid geabsorbeerd na orale toediening. Vanwege het first-pass-metabolisme was de absolute biologische beschikbaarheid echter gemiddeld 64% (44%). Er was een lineair verband tussen dosis en AUCtf (AUC van tijd nul tot de laatste meetbare concentratie) na enkelvoudige doses van 200 tot 1200 mg ribavirine. De relatie tussen dosis en Cmax was kromlijnig en neigde naar asymptoot boven enkelvoudige doses van 400 tot 600 mg.
Na meervoudige orale dosering, gebaseerd op AUC12hr, werd een 6-voudige accumulatie van ribavirine in plasma waargenomen. Na orale toediening van 600 mg tweemaal daags werd de steady-state bereikt na ongeveer 4 weken, met gemiddelde steady-state plasmaconcentraties van 2200 ng/ml (37%). Na stopzetting van de dosering was de gemiddelde halfwaardetijd 298 (30%) uur, wat waarschijnlijk een weerspiegeling is van langzame eliminatie uit niet-plasmacompartimenten.
Effect van antacidum op de absorptie van ribavirine
Gelijktijdige toediening van REBETOL-capsules met een antacidum dat magnesium, aluminium en simethicon bevat, resulteerde in een afname van 14% van de gemiddelde ribavirine-AUCtf. De klinische relevantie van de resultaten van dit onderzoek met een enkele dosis is niet bekend.
Weefselverdeling
Het transport van ribavirine naar niet-plasmacompartimenten is het meest uitgebreid bestudeerd in rode bloedcellen en er is vastgesteld dat het voornamelijk plaatsvindt via een equilibrerende nucleosidetransporter van het es-type. Dit type transporter is aanwezig op vrijwel alle celtypen en kan verantwoordelijk zijn voor het uitgebreide distributievolume. Ribavirine bindt niet aan plasma-eiwitten.
Metabolisme en uitscheiding
Ribavirine heeft twee metabolismeroutes: (i) een omkeerbare fosforyleringsroute in genucleëerde cellen; en (ii) een afbraakroute die deribosylering en amidehydrolyse omvat om een triazoolcarbonzuurmetaboliet op te leveren. Ribavirine en zijn triazoolcarboxamide en triazoolcarbonzuurmetabolieten worden via de nieren uitgescheiden. Na orale toediening van 600 mg 14C-ribavirine werd in 336 uur respectievelijk ongeveer 61% en 12% van de radioactiviteit in de urine en feces uitgescheiden. Onveranderd ribavirine was goed voor 17% van de toegediende dosis.
Speciale populaties
Nierfunctiestoornis
De farmacokinetiek van ribavirine werd beoordeeld na toediening van een enkele orale dosis (400 mg) ribavirine aan niet met HCV geïnfecteerde proefpersonen met verschillende gradaties van nierdisfunctie. De gemiddelde AUCtf-waarde was driemaal groter bij proefpersonen met een creatinineklaring tussen 10 en 30 ml/min in vergelijking met controlepersonen (creatinineklaring hoger dan 90 ml/min). Bij proefpersonen met creatinineklaringswaarden tussen 30 en 60 ml/min was de AUCtf twee keer zo hoog in vergelijking met controlepersonen. De verhoogde AUCtf lijkt het gevolg te zijn van een verlaging van de renale en niet-renale klaring bij deze proefpersonen. Fase 3-werkzaamheidsonderzoeken omvatten proefpersonen met creatinineklaringswaarden van meer dan 50 ml/min. De farmacokinetiek van meervoudige doses ribavirine kan niet nauwkeurig worden voorspeld bij patiënten met nierdisfunctie. Ribavirine wordt niet effectief verwijderd door hemodialyse.
Patiënten met een creatinineklaring van minder dan 50 ml/min mogen niet met REBETOL worden behandeld (zie: CONTRA-INDICATIES ].
Leverfunctiestoornis
Het effect van leverdisfunctie werd beoordeeld na een enkele orale dosis ribavirine (600 mg). De gemiddelde AUCtf-waarden waren niet significant verschillend bij proefpersonen met lichte, matige of ernstige leverdisfunctie (Child-Pugh-classificatie A, B of C) in vergelijking met controlepersonen. De gemiddelde Cmax-waarden namen echter toe met de ernst van de leverfunctiestoornis en waren tweemaal zo hoog bij proefpersonen met ernstige leverfunctiestoornis als bij controlepersonen.
Oudere patiënten
Farmacokinetische evaluaties bij oudere proefpersonen zijn niet uitgevoerd.
Geslacht
Er werden geen klinisch significante farmacokinetische verschillen opgemerkt in een onderzoek met een enkele dosis van 18 mannelijke en 18 vrouwelijke proefpersonen.
Pediatrische patiënten
De farmacokinetische eigenschappen van meervoudige doses van REBETOL 200 mg capsules en INTRON A bij pediatrische patiënten met chronische hepatitis C tussen 5 en 16 jaar zijn samengevat in tabel 12. De farmacokinetiek van REBETOL 200 mg en INTRON A (dosis-genormaliseerd) is vergelijkbaar bij volwassenen en pediatrische onderwerpen.
De volledige farmacokinetische kenmerken van REBETOL drank zijn niet vastgesteld bij pediatrische proefpersonen. De Cmin-waarden van ribavirine waren vergelijkbaar na toediening van REBETOL drank of REBETOL 200 mg capsules gedurende 48 weken behandeling bij pediatrische proefpersonen (3 tot 16 jaar).
Er werd een klinische studie uitgevoerd bij pediatrische proefpersonen met chronische hepatitis C tussen 3 en 17 jaar oud waarin de farmacokinetiek van PegIntron en REBETOL (capsules en drank) werd geëvalueerd. Bij pediatrische proefpersonen die een voor het lichaamsoppervlak aangepaste dosering van PegIntron van 60 mcg/m²/week kregen, werd voorspeld dat de log-getransformeerde ratio van de blootstelling tijdens het doseringsinterval 58% [90% BI: 141%, 177%] hoger zou zijn dan waargenomen bij volwassenen die 1,5 mcg/kg/week kregen. De farmacokinetiek van REBETOL (dosis-genormaliseerd) in deze studie was vergelijkbaar met die gerapporteerd in een eerdere studie van REBETOL 200 mg in combinatie met INTRON A bij pediatrische proefpersonen en bij volwassenen.
Effect van voedsel op de absorptie van ribavirine
Zowel AUCtf als Cmax namen met 70% toe wanneer REBETOL-capsules werden toegediend met een vetrijke maaltijd (841 kcal, 53,8 g vet, 31,6 g eiwit en 57,4 g koolhydraat) in een farmacokinetisch onderzoek met een enkelvoudige dosis [zie DOSERING EN ADMINISTRATIE ].
Microbiologie
Werkingsmechanisme
Het mechanisme waarmee ribavirine bijdraagt aan zijn antivirale werkzaamheid in de kliniek is niet volledig begrepen. Ribavirine heeft directe antivirale activiteit in weefselkweek tegen veel RNA-virussen. Ribavirine verhoogt de mutatiefrequentie in de genomen van verschillende virussen en ribavirinetrifosfaat remt HCV-polymerase in een biochemische reactie.
Antivirale activiteit in celcultuur
De antivirale activiteit van ribavirine in het HCV-replicon is niet goed begrepen en is niet gedefinieerd vanwege de cellulaire toxiciteit van ribavirine. Directe antivirale activiteit is waargenomen in weefselkweek van andere RNA-virussen. De anti-HCV-activiteit van interferon werd aangetoond in cellen die zelfreplicerende HCV-RNS (HCV-repliconcellen) of HCV-infectie bevatten.
Weerstand
HCV-genotypen vertonen een grote variabiliteit in hun respons op gepegyleerd recombinant humaan interferon/ribavirine-therapie. Genetische veranderingen die verband houden met de variabele respons zijn niet geïdentificeerd.
Kruisweerstand
Er is geen gemelde kruisresistentie tussen gepegyleerde/niet-gepegyleerde interferonen en ribavirine.
Dierlijke toxicologie en farmacologie
Langetermijnstudies bij muizen en ratten [18 tot 24 maanden; doses van respectievelijk 20 tot 75 en 10 tot 40 mg/kg/dag (geschatte humane equivalente doses van respectievelijk 1,67 tot 6,25 en 1,43 tot 5,71 mg/kg/dag, gebaseerd op aanpassing van het lichaamsoppervlak voor een volwassene van 60 kg; ongeveer 0,1 tot 0,4 maal de maximale humane 24-uurs dosis ribavirine)] hebben een verband aangetoond tussen chronische blootstelling aan ribavirine en verhoogde incidentie van vasculaire laesies (microscopische bloedingen) bij muizen. Bij ratten trad retinale degeneratie op bij controles, maar de incidentie was verhoogd bij met ribavirine behandelde ratten.
In een onderzoek waarin rattenjongen postnataal werden gedoseerd met ribavirine in doses van 10, 25 en 50 mg/kg/dag, traden geneesmiddelgerelateerde sterfgevallen op bij 50 mg/kg (bij plasmaconcentraties van rattenjongen lager dan de humane plasmaconcentraties bij de mens). therapeutische dosis) tussen studiedagen 13 en 48. Rattenjongen die werden gedoseerd van postnatale dag 7 tot en met 63 vertoonden een kleine, dosisgerelateerde afname van de algehele groei bij alle doses, die zich vervolgens manifesteerde als lichte afname van lichaamsgewicht, kroon-romplengte, en beenlengte. Deze effecten vertoonden aanwijzingen voor reversibiliteit en er werden geen histopathologische effecten op het bot waargenomen. Er werden geen effecten van ribavirine waargenomen met betrekking tot neurologische gedrags- of reproductieve ontwikkeling.
Klinische studies
Klinisch onderzoek 1 evalueerde PegIntron-monotherapie. Zien PegIntron-etikettering voor informatie over deze proef.
REBETOL/PegIntron-combinatietherapie
Volwassen onderwerpen
Studie 2
In een gerandomiseerde studie werd de behandeling vergeleken met twee PegIntron/REBETOL 200 mg-regimes [PegIntron 1,5 mcg/kg subcutaan eenmaal per week/REBETOL 800 mg oraal per dag (in verdeelde doses); PegIntron 1,5 mcg/kg subcutaan eenmaal per week gedurende 4 weken daarna 0,5 mcg/kg subcutaan eenmaal per week gedurende 44 weken/REBETOL 1000 of 1200 mg oraal per dag (in verdeelde doses)] met INTRON A [3 MIU subcutaan driemaal per week/REBETOL 1000 of 1200 mg oraal per dag (in verdeelde doses)] bij 1530 volwassenen met chronische hepatitis C. Interferon-naïeve proefpersonen werden 48 weken behandeld en 24 weken na de behandeling gevolgd. In aanmerking komende proefpersonen hadden gecompenseerde leverziekte, detecteerbaar HCV-RNA, verhoogd ALT en leverhistopathologie consistent met chronische hepatitis.
De respons op de behandeling werd 24 weken na de behandeling gedefinieerd als niet-detecteerbaar HCV-RNA (zie tabel 13). Het responspercentage op de dosis PegIntron 1,5 mcg/kg en ribavirine 800 mg was hoger dan het responspercentage op INTRON A/REBETOL (zie tabel 13). Het responspercentage op PegIntron 1,5 → 0,5 mcg/kg/REBETOL 200 mg was in wezen hetzelfde als het antwoord op INTRON A/REBETOL (gegevens niet getoond).
Proefpersonen met viraal genotype 1, ongeacht de virale belasting, hadden een lagere respons op PegIntron (1,5 mcg/kg)/REBETOL (800 mg) in vergelijking met proefpersonen met andere virale genotypen. Proefpersonen met beide slechte prognostische factoren (genotype 1 en hoge virale belasting) hadden een responspercentage van 30% (78/256) vergeleken met een responspercentage van 29% (71/247) met de combinatietherapie INTRON A/REBETOL.
Proefpersonen met een lager lichaamsgewicht hadden over het algemeen een hoger percentage bijwerkingen [zie: ONGEWENSTE REACTIES en hogere responspercentages dan proefpersonen met een hoger lichaamsgewicht. Verschillen in responspercentages tussen behandelarmen varieerden niet substantieel met het lichaamsgewicht.
De responspercentages op de behandeling met PegIntron/REBETOL-combinatietherapie waren 49% bij mannen en 56% bij vrouwen. De responspercentages waren lager bij Afro-Amerikaanse en Spaanse proefpersonen en hoger bij Aziaten in vergelijking met blanken. Hoewel Afro-Amerikanen een groter aandeel slechte prognostische factoren hadden in vergelijking met blanken, was het aantal niet-blanken dat werd bestudeerd (11% van het totaal) onvoldoende om zinvolle conclusies te kunnen trekken over verschillen in responspercentages na correctie voor prognostische factoren in dit onderzoek.
Bij 68% van de proefpersonen werden voor en na de behandeling leverbiopten verkregen. Vergeleken met de uitgangswaarde werd bij ongeveer tweederde van de proefpersonen in alle behandelingsgroepen een bescheiden vermindering van de ontsteking waargenomen.
Studie 3
In een groot gemeenschapsonderzoek in de Verenigde Staten werden 4913 proefpersonen met chronische hepatitis C gerandomiseerd om PegIntron 1,5 mcg/kg eenmaal per week subcutaan te krijgen in combinatie met een REBETOL 200 mg dosis van 800 tot 1400 mg (gewichtsgebaseerde dosering [WBD]) of 800 mg (plat) oraal per dag (in verdeelde doses) gedurende 24 of 48 weken op basis van genotype. De respons op de behandeling werd 24 weken na de behandeling gedefinieerd als niet-detecteerbaar HCV-RNA (gebaseerd op een test met een ondergrens van detectie van 125 IE/ml).
Behandeling met PegIntron 1,5 mcg/kg en REBETOL 800 tot 1400 mg resulteerde in een hogere aanhoudende virologische respons in vergelijking met PegIntron in combinatie met een vaste dagelijkse dosis van 800 mg REBETOL. Proefpersonen die meer dan 105 kg wogen, behaalden het grootste voordeel met WBD, hoewel een bescheiden voordeel ook werd waargenomen bij proefpersonen die meer dan 85 tot 105 kg wogen (zie tabel 14). Het voordeel van WBD bij proefpersonen die meer dan 85 kg wogen, werd waargenomen met HCV-genotypen 1-3. Er waren onvoldoende gegevens beschikbaar om conclusies te trekken over andere genotypen. Het gebruik van WBD resulteerde in een verhoogde incidentie van bloedarmoede [zie: ONGEWENSTE REACTIES ].
Een totaal van 1552 proefpersonen die meer dan 65 kg wogen in onderzoek 3 hadden genotype 2 of 3 en werden gerandomiseerd naar behandeling van 24 of 48 weken. Er werd geen bijkomend voordeel waargenomen bij een langere behandelingsduur.
Studie 4
Een groot gerandomiseerd onderzoek vergeleek de veiligheid en werkzaamheid van een behandeling gedurende 48 weken met twee PegIntron/REBETOL 200 mg-regimes [PegIntron 1,5 mcg/kg en 1 mcg/kg subcutaan eenmaal per week, beide in combinatie met REBETOL 800 tot 1400 mg PO per dag (in twee verdeelde doses)] en Pegasys 180 mcg subcutaan eenmaal per week in combinatie met Copegus 1000 tot 1200 mg PO per dag (in twee verdeelde doses) bij 3070 nog niet eerder behandelde volwassenen met chronisch hepatitis C-genotype 1. In dit onderzoek ontbrak een vroege virologische respons (niet-detecteerbaar HCV-RNA of meer dan of gelijk aan 2 log10 reductie ten opzichte van baseline) in behandelingsweek 12 was het criterium voor stopzetting van de behandeling. SVR werd 24 weken na de behandeling gedefinieerd als niet-detecteerbaar HCV-RNA (Roche COBAS TaqMan-assay, een ondergrens van kwantificering van 27 IE/ml) (zie tabel 15).
De totale SVR-percentages waren vergelijkbaar tussen de drie behandelingsgroepen. Ongeacht de behandelingsgroep waren de SVR-percentages lager bij proefpersonen met slechte prognostische factoren. Proefpersonen met slechte prognostische factoren gerandomiseerd naar PegIntron (1,5 mcg/kg)/REBETOL of Pegasys/Copegus bereikten echter hogere SVR-percentages in vergelijking met vergelijkbare proefpersonen die waren gerandomiseerd naar PegIntron 1 mcg/kg/REBETOL. Voor de dosis PegIntron 1,5 mcg/kg en REBETOL waren de SVR-percentages voor proefpersonen met en zonder de volgende prognostische factoren als volgt: cirrose (10% vs. 42%), normale ALT-waarden (32% vs. 42%), virale baseline belasting van meer dan 600.000 IE/ml (35% vs. 61%), 40 jaar en ouder (38% vs. 50%) en Afro-Amerikaans ras (23% vs. 44%). Bij proefpersonen met niet-detecteerbaar HCV-RNA in behandelingsweek 12 die PegIntron (1,5 mcg/kg)/REBETOL 200 mg kregen, was het SVR-percentage 81% (328/407).
Onderzoek 5 - REBETOL/PegIntron-combinatietherapie bij eerdere mislukte behandelingen
In een niet-vergelijkend onderzoek werden 2293 proefpersonen met matige tot ernstige fibrose bij wie eerdere behandeling met combinatie alfa-interferon/ribavirine niet had gefaald, opnieuw behandeld met PegIntron, 1,5 mcg/kg subcutaan, eenmaal per week, in combinatie met ribavirine aangepast aan het gewicht. In aanmerking komende proefpersonen waren onder meer eerdere non-responders (patiënten die HCV-RNA-positief waren aan het einde van een behandeling van minimaal 12 weken) en eerdere recidiverende personen (patiënten die HCVRNA-negatief waren aan het einde van een behandeling van minimaal 12 weken en daarna een terugval kregen na de behandeling opvolgen). Proefpersonen die in week 12 negatief waren, werden 48 weken behandeld en 24 weken na de behandeling gevolgd. De respons op de behandeling werd gedefinieerd als niet-detecteerbaar HCV-RNA 24 weken na de behandeling (gemeten met een op onderzoek gebaseerde test, detectielimiet 125 IE/ml). Het totale responspercentage was 22% (497/2293) (99% BI: 19,5; 23,9). Proefpersonen met de volgende kenmerken hadden minder kans op herbehandeling: eerdere non-respons, eerdere behandeling met gepegyleerd interferon, significante overbruggende fibrose of cirrose en genotype 1-infectie.
De aanhoudende virologische responspercentages bij herbehandeling volgens baselinekenmerken zijn samengevat in Tabel 16.
Het bereiken van een niet-detecteerbaar HCV-RNA in behandelingsweek 12 was een sterke voorspeller van SVR. In dit onderzoek bereikten 1470 (64%) proefpersonen geen niet-detecteerbaar HCV-RNA in behandelingsweek 12 en werd deelname aan langetermijnbehandelingsonderzoeken aangeboden vanwege een ontoereikende respons op de behandeling. Van de 823 (36%) proefpersonen bij wie HCV-RNA niet detecteerbaar was in behandelingsweek 12, hadden degenen die geïnfecteerd waren met genotype 1 een SVR van 48% (245/507), met een reeks reacties op fibrosescores (F4-F2) van 39-55%. Proefpersonen die geïnfecteerd waren met genotype 2/3 en die HCV-RNA niet detecteerbaar waren in behandelingsweek 12, hadden een totale SVR van 70% (196/281), met een reeks responsen volgens fibrosescores (F4-F2) van 60-83%. Voor alle genotypen waren hogere fibrosescores geassocieerd met een verminderde kans op het bereiken van SVR.
Pediatrische onderwerpen
Eerder onbehandelde pediatrische proefpersonen van 3 tot 17 jaar met gecompenseerde chronische hepatitis C en detecteerbaar HCV-RNA werden behandeld met REBETOL 15 mg/kg per dag en PegIntron 60 mcg/m² eenmaal per week gedurende 24 of 48 weken op basis van HCV-genotype en virale baseline laden. Alle proefpersonen moesten 24 weken na de behandeling worden gevolgd. In totaal werden 107 proefpersonen behandeld, waarvan 52% vrouw, 89% blank en 67% geïnfecteerd met HCV-genotype 1. Personen geïnfecteerd met genotypes 1, 4 of genotype 3 met HCV-RNA groter dan of gelijk aan 600.000 IE/ml kregen 48 weken therapie, terwijl degenen die geïnfecteerd waren met genotype 2 of genotype 3 met HCV-RNA van minder dan 600.000 IE/ml 24 weken therapie kregen. De onderzoeksresultaten zijn samengevat in Tabel 17.
REBETOL/INTRON Een combinatietherapie
Volwassen onderwerpen
Eerder onbehandelde onderwerpen
Volwassenen met gecompenseerde chronische hepatitis C en detecteerbaar HCV-RNA (beoordeeld door een centraal laboratorium met behulp van een op onderzoek gebaseerde RT-PCR-test) die niet eerder behandeld waren met alfa-interferontherapie, werden opgenomen in twee dubbelblinde multicenteronderzoeken (VS en internationaal) en gerandomiseerd om REBETOL 200 mg capsules 1200 mg/dag (1000 mg/dag voor personen met een gewicht van minder dan of gelijk aan 75 kg) en INTRON A 3 MIU driemaal per week of INTRON A en placebo gedurende 24 of 48 weken te ontvangen, gevolgd door 24 weken van follow-up buiten de therapie. De internationale studie bevatte geen behandelarm met INTRON A en placebo van 24 weken. Aan het Amerikaanse onderzoek namen 912 proefpersonen deel die bij baseline 67% mannelijk waren, 89% blank met een gemiddelde Knodell HAI-score (I+II+III) van 7,5 en 72% genotype 1. Het internationale onderzoek, uitgevoerd in Europa, Israël , Canada en Australië, namen 799 proefpersonen op (65% mannelijk, 95% blank, gemiddelde Knodell-score 6,8 en 58% genotype 1).
De onderzoeksresultaten zijn samengevat in Tabel 18.
Van de proefpersonen die in week 24 van de behandeling met REBETOL/INTRON A geen HCV-RNA hadden bereikt onder de detectielimiet van de op onderzoek gebaseerde test, reageerde minder dan 5% op een aanvullende combinatiebehandeling van 24 weken.
Van de proefpersonen met HCV-genotype 1 die werden behandeld met REBETOL/INTRON A-therapie en die HCV-RNA bereikten onder de detectielimiet van de op onderzoek gebaseerde test na 24 weken, hadden degenen die waren gerandomiseerd naar 48 weken behandeling een hogere virologische respons in vergelijking met die in de 24- week behandelgroep. Er werd geen toename in responspercentages waargenomen bij proefpersonen met HCV niet-genotype 1 gerandomiseerd naar behandeling met REBETOL/INTRON A gedurende 48 weken in vergelijking met 24 weken.
Terugval onderwerpen
Proefpersonen met gecompenseerde chronische hepatitis C en detecteerbaar HCV-RNA (beoordeeld door een centraal laboratorium met behulp van een op onderzoek gebaseerde RT-PCR-assay) die een recidief hadden gekregen na een of twee interferontherapieën (gedefinieerd als abnormale serum-ALAT-spiegels) werden opgenomen in twee multicenter, dubbelblinde onderzoeken (VS en internationaal) en gerandomiseerd om REBETOL 1200 mg/dag (1000 mg/dag voor proefpersonen met een gewicht 75 kg) en INTRON A 3 MIU driemaal per week of INTRON A en placebo gedurende 24 weken te krijgen, gevolgd door 24 weken off-therapie follow-up. Aan het Amerikaanse onderzoek namen 153 proefpersonen deel die bij baseline 67% mannelijk waren, 92% blank met een gemiddelde Knodell HAI-score (I+II+III) van 6,8 en 58% genotype 1. Het internationale onderzoek, uitgevoerd in Europa, Israël , Canada en Australië, schreven 192 proefpersonen in (64% mannelijk, 95% blank, gemiddelde Knodell-score 6,6 en 56% genotype 1). De onderzoeksresultaten zijn samengevat in Tabel 19.
Virologische en histologische reacties waren vergelijkbaar bij mannelijke en vrouwelijke proefpersonen in zowel de eerder onbehandelde als de recidiefonderzoeken.
Pediatrische onderwerpen
Pediatrische proefpersonen van 3 tot 16 jaar met gecompenseerde chronische hepatitis C en detecteerbaar HCV-RNA (beoordeeld door een centraal laboratorium met behulp van een op onderzoek gebaseerde RT-PCR-assay) werden behandeld met REBETOL 15 mg/kg per dag en INTRON A 3 MIU/ m² driemaal per week gedurende 48 weken gevolgd door 24 weken off-therapie follow-up. In totaal werden 118 proefpersonen behandeld, waarvan 57% man, 80% blank en 78% genotype 1. Proefpersonen jonger dan 5 jaar kregen REBETOL drank en degenen van 5 jaar of ouder kregen ofwel REBETOL drank of capsules.
De onderzoeksresultaten zijn samengevat in Tabel 20.
Proefpersonen met viraal genotype 1, ongeacht de viral load, hadden een lager responspercentage op INTRON A/REBETOL-combinatietherapie in vergelijking met proefpersonen met genotype non-1, 36% versus 81%. Proefpersonen met beide slechte prognostische factoren (genotype 1 en hoge viral load) hadden een respons van 26% (13/50).
PATIËNT INFORMATIE
Geen informatie verstrekt. Raadpleeg de WAARSCHUWINGEN EN VOORZORGSMAATREGELEN sectie.