Epivir 150mg Lamivudine Gebruik, bijwerkingen en dosering. Prijs in online apotheek. Generieke medicijnen zonder recept.
Wat is Epivir en hoe wordt het gebruikt?
Epivir 150 mg is een receptgeneesmiddel dat wordt gebruikt om de symptomen van hiv-infectie en chronische hepatitis B te behandelen. Epivir 150 mg kan alleen of in combinatie met andere medicijnen worden gebruikt.
Epivir behoort tot een klasse geneesmiddelen die Hepatitis B/Hepatitis C-agentia worden genoemd; HIV, NRTI's.
Het is niet bekend of Epivir veilig en effectief is bij kinderen jonger dan 3 maanden.
Wat zijn de mogelijke bijwerkingen van Epivir 150 mg?
Epivir kan ernstige bijwerkingen veroorzaken, waaronder:
- netelroos,
- moeite met ademhalen,
- zwelling van uw gezicht, lippen, tong of keel,
- ongebruikelijke spierpijn,
- buikpijn,
- braken,
- onregelmatige hartslag,
- duizeligheid,
- koud hebben,
- zwakheid,
- vermoeidheid,
- hevige pijn in uw bovenbuik die zich uitbreidt naar uw rug,
- misselijkheid,
- snelle hartslag,
- zwelling rond uw buik,
- rechtszijdige pijn in de bovenbuik,
- verlies van eetlust,
- donkere urine,
- kleikleurige ontlasting,
- geel worden van de huid of ogen (geelzucht),
- koorts,
- Nacht zweet,
- opgezwollen klieren,
- koortsblaasjes,
- hoesten,
- piepende ademhaling,
- diarree,
- gewichtsverlies,
- moeite met spreken of slikken,
- problemen met evenwicht of oogbewegingen,
- stekelig gevoel,
- zwelling in uw nek of keel (vergrote schildklier),
- menstruatieveranderingen, en
- impotentie
Roep meteen medische hulp in als u een van de bovenstaande symptomen heeft.
De meest voorkomende bijwerkingen van Epivir zijn:
- misselijkheid,
- diarree,
- hoofdpijn,
- koorts,
- vermoeidheid,
- algemeen ziek gevoel,
- oorpijn of vol gevoel,
- moeite met horen,
- drainage uit het oor,
- onrust bij een kind,
- verstopte neus,
- niezen,
- keelpijn, en
- hoesten
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van Epivir. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
WAARSCHUWING
EXACERBATIES VAN HEPATITIS B, en VERSCHILLENDE FORMULERINGEN VAN EPIVIR.
Exacerbaties van hepatitis B
Ernstige acute exacerbaties van hepatitis B zijn gemeld bij patiënten die gelijktijdig zijn geïnfecteerd met het hepatitis B-virus (HBV) en het humaan immunodeficiëntievirus (hiv-1) en die zijn gestopt met EPIVIR. Bij patiënten die stoppen met EPIVIR en die gelijktijdig zijn geïnfecteerd met HIV-1 en HBV, moet de leverfunctie nauwlettend worden gecontroleerd met zowel klinische als laboratoriumfollow-up gedurende ten minste enkele maanden. Indien van toepassing kan het starten van anti-hepatitis B-therapie gerechtvaardigd zijn (zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ).
Belangrijke verschillen tussen producten die lamivudine bevatten
EPIVIR 150 mg tabletten en drank (gebruikt voor de behandeling van HIV-1-infectie) bevatten een hogere dosis van het werkzame bestanddeel (lamivudine) dan EPIVIR-HBV-tabletten en drank (gebruikt voor de behandeling van chronische HBV-infectie). Patiënten met een HIV-1-infectie mogen alleen doseringsvormen krijgen die geschikt zijn voor de behandeling van HIV-1 (zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ).
OMSCHRIJVING
EPIVIR (ook bekend als 3TC) is een merknaam voor lamivudine, een synthetisch nucleoside-analoog met activiteit tegen HIV-1 en HBV. De chemische naam van lamivudine is (2R,cis)-4-amino-1(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidine-2-on. Lamivudine is de (-)enantiomeer van een dideoxy-analoog van cytidine. Lamivudine wordt ook wel (-)2',3'-dideoxy, 3'thiacytidine genoemd. Het heeft een molecuulformule van C8H11N3O3S en een molecuulgewicht van 229,3 g per mol. Het heeft de volgende structuurformule:
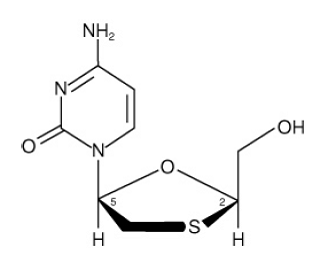
Lamivudine is een witte tot gebroken witte kristallijne vaste stof met een oplosbaarheid van ongeveer 70 mg per ml in water bij 20°C.
EPIVIR 150 mg tabletten zijn voor orale toediening. Elke filmomhulde tablet van 150 mg bevat 150 mg lamivudine en de inactieve ingrediënten hypromellose, magnesiumstearaat, microkristallijne cellulose, polyethyleenglycol, polysorbaat 80, natriumzetmeelglycolaat en titaniumdioxide.
Elke filmomhulde tablet van 300 mg bevat 300 mg lamivudine en de inactieve ingrediënten zwart ijzeroxide, hypromellose, magnesiumstearaat, microkristallijne cellulose, polyethyleenglycol, polysorbaat 80, natriumzetmeelglycolaat en titaniumdioxide.
EPIVIR drank is voor orale toediening. Eén milliliter (1 ml) EPIVIR drank bevat 10 mg lamivudine (10 mg per ml) in een waterige oplossing en de inactieve ingrediënten kunstmatige aardbeien- en bananensmaakstoffen, citroenzuur (watervrij), methylparaben, propyleenglycol, propylparaben, natriumcitraat (dihydraat) en sucrose (200 mg).
INDICATIES
EPIVIR 150 mg is een nucleoside-analoog die is geïndiceerd in combinatie met andere antiretrovirale middelen voor de behandeling van infectie met het humaan immunodeficiëntievirus type 1 (hiv-1).
Beperkingen van gebruik
- De dosering van dit product is voor HIV-1 en niet voor HBV.
DOSERING EN ADMINISTRATIE
Aanbevolen dosering voor volwassen patiënten
De aanbevolen dosering van EPIVIR 150 mg bij met HIV-1 geïnfecteerde volwassenen is 300 mg per dag, toegediend als 150 mg tweemaal daags oraal of 300 mg eenmaal daags oraal met of zonder voedsel. Als lamivudine wordt toegediend aan een patiënt die is geïnfecteerd met hiv-1 en HBV, moet de dosering die is aangegeven voor hiv-1-therapie worden gebruikt als onderdeel van een geschikt combinatieschema [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Aanbevolen dosering voor pediatrische patiënten
EPIVIR 150 mg tablet met breukstreep is de voorkeursformulering voor met HIV-1 geïnfecteerde pediatrische patiënten die ten minste 14 kg wegen en voor wie een vaste doseringsvorm geschikt is. Alvorens EPIVIR-tabletten met breukstreep voor te schrijven, moeten pediatrische patiënten worden beoordeeld op het vermogen om tabletten door te slikken. Voor patiënten die niet in staat zijn om EPIVIR 150 mg tabletten veilig en betrouwbaar door te slikken, kan de formulering voor drank worden voorgeschreven [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. De aanbevolen orale dosering van EPIVIR 150 mg tabletten voor met HIV-1 geïnfecteerde pediatrische patiënten wordt weergegeven in Tabel 1.
Orale oplossing
De aanbevolen dosering van EPIVIR 150 mg drank bij met HIV-1 geïnfecteerde pediatrische patiënten van 3 maanden en ouder is 5 mg per kg tweemaal daags oraal of 10 mg per kg eenmaal daags oraal (tot een maximum van 300 mg per dag), toegediend in combinatie met andere antiretrovirale middelen [zie: KLINISCHE FARMACOLOGIE ]. Houd rekening met de virale last van HIV-1 en het aantal CD4+-cellen/percentages bij het selecteren van het doseringsinterval voor patiënten die beginnen met de behandeling met drank [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , KLINISCHE FARMACOLOGIE ].
Patiënten met nierinsufficiëntie
De dosering van EPIVIR 150 mg wordt aangepast aan de nierfunctie. Doseringsaanpassingen staan vermeld in Tabel 2 [zie KLINISCHE FARMACOLOGIE ].
Er is geen extra dosering van EPIVIR 150 mg nodig na routinematige (4 uur durende) hemodialyse of peritoneale dialyse.
Hoewel er onvoldoende gegevens zijn om een specifieke dosisaanpassing van EPIVIR aan te bevelen bij pediatrische patiënten met een nierfunctiestoornis, moet een verlaging van de dosis en/of een verhoging van het doseringsinterval worden overwogen.
HOE GELEVERD
Doseringsvormen en sterke punten
EPIVIR gescoorde tabletten
EPIVIR-tabletten met breukstreep bevatten 150 mg lamivudine. De tabletten zijn witte, ruitvormige, filmomhulde tabletten met een breukgleuf en aan beide zijden gegraveerd met “GX CJ7”.
EPIVIR-tabletten
EPIVIR-tabletten bevatten 300 mg lamivudine. De tabletten zijn grijs, gemodificeerd ruitvormig, filmomhuld en gegraveerd met “GX EJ7” aan de ene kant en glad aan de andere kant.
EPIVIR orale oplossing
EPIVIR drank bevat 10 mg lamivudine per 1 ml. De oplossing is een heldere, kleurloze tot lichtgele vloeistof met aardbeiensmaak.
Opslag en behandeling
EPIVIR Tabletten met breukstreep bevatten 150 mg lamivudine, zijn wit, ruitvormig, filmomhuld en aan beide zijden bedrukt met “GX CJ7”. Als volgt verpakt:
Fles van 60 tabletten ( NDC 49702-203-18) met kindveilige sluiting.
EPIVIR tabletten bevatten 300 mg lamivudine, zijn grijs, gemodificeerd ruitvormig, filmomhuld en gegraveerd met “GX EJ7” aan de ene kant en glad aan de andere kant. Als volgt verpakt:
Fles van 30 tabletten ( NDC 49702-204-13) met kindveilige sluiting.
Aanbevolen opslag
Bewaar EPIVIR-tabletten bij 25°C (77°F); excursies toegestaan tot 15° tot 30°C (59° tot 86°F) [zie USP-gecontroleerde kamertemperatuur ].
EPIVIR drank is een heldere, kleurloze tot lichtgele vloeistof met aardbeiensmaak. Elke ml van de oplossing bevat 10 mg lamivudine. Als volgt verpakt:
Plastic fles van 240 ml ( NDC 49702-205-48) met kindveilige sluiting. Dit product vereist geen reconstitutie.
Bewaren in goed gesloten flessen bij 25°C (77°F) [zie USP-gecontroleerde kamertemperatuur ].
Gefabriceerd voor: ViiV Healthcare Research Triangle Park, NC 27709. door: GlaxoSmithKline Research Triangle Park, NC 27709, Gefabriceerd in overeenstemming met Shire Pharmaceuticals Group plc. Herzien: april 2018
BIJWERKINGEN
De volgende bijwerkingen worden besproken in andere secties van de etikettering:
- Exacerbaties van hepatitis B [zie GEVAARLIJKE WAARSCHUWING: , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
- Lactaatacidose en ernstige hepatomegalie met steatose [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
- Leverdecompensatie bij patiënten met gelijktijdige infectie met HIV-1 en hepatitis C [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
- Pancreatitis [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
- Immuunreconstitutiesyndroom [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Ervaring met klinische proeven
Ervaring met klinische proeven bij volwassen proefpersonen
Omdat klinische onderzoeken onder sterk uiteenlopende omstandigheden worden uitgevoerd, kunnen de bijwerkingen die in de klinische onderzoeken van een geneesmiddel zijn waargenomen niet direct worden vergeleken met de percentages in de klinische onderzoeken van een ander geneesmiddel en komen mogelijk niet overeen met de percentages die in de klinische praktijk worden waargenomen.
Het veiligheidsprofiel van EPIVIR 150 mg bij volwassenen is voornamelijk gebaseerd op 3.568 met hiv-1 geïnfecteerde proefpersonen in 7 klinische onderzoeken.
De meest voorkomende bijwerkingen zijn hoofdpijn, misselijkheid, malaise, vermoeidheid, nasale tekenen en symptomen, diarree en hoesten.
Geselecteerde klinische bijwerkingen bij meer dan of gelijk aan 5% van de proefpersonen tijdens behandeling met EPIVIR 150 mg tweemaal daags plus RETROVIR 200 mg 3 maal daags gedurende maximaal 24 weken worden vermeld in Tabel 3.
Pancreatitis
Pancreatitis werd waargenomen bij 9 van de 2.613 volwassen proefpersonen (0,3%) die EPIVIR 150 mg kregen in gecontroleerde klinische onderzoeken EPV20001, NUCA3001, NUCB3001, NUCA3002, NUCB3002 en NUCB3007 [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
EPIVIR 300 mg eenmaal daags
De typen en frequenties van klinische bijwerkingen die zijn gemeld bij proefpersonen die gedurende 48 weken eenmaal daags 300 mg EPIVIR of 150 mg EPIVIR tweemaal daags (in combinatieregimes met 3 geneesmiddelen in EPV20001 en EPV40001) gedurende 48 weken kregen, waren vergelijkbaar.
Geselecteerde laboratoriumafwijkingen die tijdens de therapie zijn waargenomen, zijn samengevat in Tabel 4.
De frequenties van geselecteerde laboratoriumafwijkingen die werden gemeld bij proefpersonen die EPIVIR 300 mg eenmaal daags of EPIVIR 150 mg tweemaal daags kregen (in combinatieregimes met 3 geneesmiddelen in EPV20001 en EPV40001) waren vergelijkbaar.
Ervaring met klinische onderzoeken bij pediatrische proefpersonen
EPIVIR drank is onderzocht bij 638 pediatrische proefpersonen in de leeftijd van 3 maanden tot 18 jaar in 3 klinische onderzoeken.
Geselecteerde klinische bijwerkingen en fysieke bevindingen met een frequentie van meer dan of gelijk aan 5% tijdens behandeling met EPIVIR 4 mg per kg tweemaal daags plus RETROVIR 160 mg per m² 3 maal daags bij therapie-naïeve (minder dan of gelijk aan 56 dagen antiretrovirale therapie) pediatrische proefpersonen worden vermeld in Tabel 5.
Pancreatitis
Pancreatitis, die in sommige gevallen fataal was, is waargenomen bij pediatrische patiënten die eerder met antiretrovirale nucleoside zijn behandeld en die EPIVIR alleen of in combinatie met andere antiretrovirale middelen kregen. In een open-label dosis-escalatie-onderzoek (NUCA2002) ontwikkelden 14 proefpersonen (14%) pancreatitis terwijl ze monotherapie met EPIVIR kregen. Drie van deze proefpersonen stierven aan complicaties van pancreatitis. In een tweede open-label onderzoek (NUCA2005) ontwikkelden 12 proefpersonen (18%) pancreatitis. In Trial ACTG300 werd pancreatitis niet waargenomen bij 236 proefpersonen die waren gerandomiseerd naar EPIVIR 150 mg plus RETROVIR. Pancreatitis werd waargenomen bij 1 proefpersoon in dit onderzoek die open-label EPIVIR kreeg in combinatie met RETROVIR en ritonavir na stopzetting van de monotherapie met didanosine (zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Paresthesieën en perifere neuropathie
Paresthesieën en perifere neuropathieën werden gemeld bij 15 proefpersonen (15%) in proef NUCA2002, 6 proefpersonen (9%) in proef NUCA2005 en 2 proefpersonen (minder dan 1%) in proef ACTG300.
Geselecteerde laboratoriumafwijkingen die therapienaïeve (minder dan of gelijk aan 56 dagen antiretrovirale therapie) pediatrische patiënten ondervonden, worden vermeld in Tabel 6.
Pediatrische patiënten eenmaal daags versus tweemaal daags dosering (COL105677)
De veiligheid van eenmaal daagse dosering vergeleken met tweemaal daagse dosering van EPIVIR 150 mg werd beoordeeld in het ARROW-onderzoek. De primaire veiligheidsbeoordeling in het ARROW-onderzoek was gebaseerd op bijwerkingen van graad 3 en graad 4. De frequentie van bijwerkingen van graad 3 en 4 was vergelijkbaar bij proefpersonen die waren gerandomiseerd naar eenmaal daagse dosering vergeleken met proefpersonen die waren gerandomiseerd naar tweemaal daagse dosering. Eén voorval van graad 4 hepatitis in het eenmaal daagse cohort werd door de onderzoeker als onzekere causaliteit beschouwd en alle andere graad 3 of 4 bijwerkingen werden door de onderzoeker als niet gerelateerd beschouwd.
pasgeborenen
Er is beperkte veiligheidsinformatie op korte termijn beschikbaar uit 2 kleine, ongecontroleerde onderzoeken in Zuid-Afrika bij pasgeborenen die lamivudine met of zonder zidovudine kregen gedurende de eerste levensweek na maternale behandeling vanaf week 38 of 36 van de zwangerschap [zie KLINISCHE FARMACOLOGIE ]. Geselecteerde bijwerkingen die bij deze pasgeborenen werden gemeld, waren onder meer verhoogde leverfunctietesten, bloedarmoede, diarree, elektrolytenstoornissen, hypoglykemie, geelzucht en hepatomegalie, huiduitslag, luchtweginfecties en sepsis; 3 pasgeborenen stierven (1 van gastro-enteritis met acidose en convulsies, 1 van traumatisch letsel en 1 van onbekende oorzaken). Er werden twee andere niet-fatale gevallen van gastro-enteritis of diarree gemeld, waarvan 1 met convulsies; 1 zuigeling had voorbijgaande nierinsufficiëntie geassocieerd met uitdroging. De afwezigheid van controlegroepen beperkt de beoordeling van de causaliteit, maar er moet worden aangenomen dat perinataal blootgestelde zuigelingen een risico kunnen lopen op bijwerkingen die vergelijkbaar zijn met de bijwerkingen die zijn gemeld bij pediatrische en volwassen met hiv-1 geïnfecteerde patiënten die worden behandeld met lamivudine-bevattende combinatieregimes. De langetermijneffecten van blootstelling aan lamivudine in utero en zuigelingen zijn niet bekend.
Postmarketingervaring
De volgende bijwerkingen zijn vastgesteld tijdens het gebruik van EPIVIR na goedkeuring. Omdat deze reacties vrijwillig worden gemeld door een populatie van onbekende grootte, is het niet altijd mogelijk om een betrouwbare schatting van de frequentie ervan of een oorzakelijk verband met blootstelling aan geneesmiddelen vast te stellen. Deze reacties zijn gekozen voor opname vanwege een combinatie van hun ernst, frequentie van melding of mogelijk causaal verband met lamivudine.
Lichaam als geheel
Herverdeling/ophoping van lichaamsvet.
Endocrien en metabool
Hyperglykemie.
Algemeen
Zwakheid. Hemische en lymfatische anemie (inclusief aplasie van zuivere rode bloedcellen en ernstige anemieën die voortschrijden tijdens de therapie).
Lever en pancreas
Melkzuuracidose en leversteatose [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ], exacerbaties van hepatitis B na de behandeling [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
overgevoeligheid
Anafylaxie, urticaria.
Musculoskeletaal
Spierzwakte, CPK-verhoging, rabdomyolyse. Huid Alopecia, jeuk.
DRUG-INTERACTIES
Geneesmiddelen die organische kationtransporters remmen
Lamivudine wordt voornamelijk in de urine geëlimineerd door actieve organische kationische secretie. De mogelijkheid van interacties met andere gelijktijdig toegediende geneesmiddelen moet worden overwogen, vooral wanneer hun belangrijkste eliminatieroute actieve renale secretie is via het organische kationische transportsysteem (bijv. trimethoprim) [zie KLINISCHE FARMACOLOGIE ]. Er zijn geen gegevens beschikbaar over interacties met andere geneesmiddelen die een nierklaringsmechanisme hebben dat vergelijkbaar is met dat van lamivudine.
Sorbitol
Gelijktijdige toediening van enkelvoudige doses lamivudine en sorbitol resulteerde in een dosisafhankelijke verlaging van de blootstelling aan lamivudine op basis van sorbitol. Vermijd indien mogelijk het gebruik van sorbitolbevattende geneesmiddelen met lamivudine [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , KLINISCHE FARMACOLOGIE ].
WAARSCHUWINGEN
Inbegrepen als onderdeel van de PREVENTIEVE MAATREGELEN sectie.
PREVENTIEVE MAATREGELEN
Patiënten met gelijktijdige infectie met hepatitis B-virus
Exacerbaties van hepatitis na de behandeling
Klinische en laboratoriumgegevens van exacerbaties van hepatitis zijn opgetreden na stopzetting van lamivudine. Deze exacerbaties zijn voornamelijk gedetecteerd door serum-ALAT-verhogingen naast het opnieuw opduiken van HBV-DNA. Hoewel de meeste voorvallen zelfbeperkend lijken te zijn, zijn er in sommige gevallen gevallen met dodelijke afloop gemeld. Soortgelijke gebeurtenissen zijn gemeld uit postmarketingervaring na veranderingen van lamivudine-bevattende hiv-1-behandelingsregimes naar niet-lamivudine-bevattende regimes bij patiënten die geïnfecteerd zijn met zowel hiv-1 als HBV. Het oorzakelijk verband met het staken van de behandeling met lamivudine is niet bekend. Patiënten moeten gedurende ten minste enkele maanden na het stoppen van de behandeling nauwlettend worden gevolgd met zowel klinische als laboratoriumfollow-up.
Belangrijke verschillen tussen producten die lamivudine bevatten
EPIVIR 150 mg tabletten en drank bevatten een hogere dosis van hetzelfde werkzame bestanddeel (lamivudine) dan EPIVIR-HBV tabletten en EPIVIR-HBV drank. EPIVIR-HBV is ontwikkeld voor patiënten met chronische hepatitis B. De formulering en dosering van lamivudine in EPIVIR-HBV zijn niet geschikt voor patiënten met gelijktijdige infectie met HIV-1 en HBV. De veiligheid en werkzaamheid van lamivudine voor de behandeling van chronische hepatitis B bij patiënten met gelijktijdige infectie met HIV-1 en HBV zijn niet vastgesteld. Als behandeling met EPIVIR-HBV wordt voorgeschreven voor chronische hepatitis B aan een patiënt met een niet-herkende of onbehandelde hiv-1-infectie, is het waarschijnlijk dat er snel hiv-1-resistentie ontstaat vanwege de subtherapeutische dosis en de ongeschiktheid van monotherapie hiv-1-behandeling. Als wordt besloten om lamivudine toe te dienen aan patiënten die gelijktijdig zijn geïnfecteerd met HIV-1 en HBV, moeten EPIVIR 150 mg tabletten, EPIVIR 150 mg drank of een ander product dat de hogere dosis lamivudine bevat, worden gebruikt als onderdeel van een geschikt combinatieregime.
Opkomst van lamivudine-resistente HBV
De veiligheid en werkzaamheid van lamivudine zijn niet vastgesteld voor de behandeling van chronische hepatitis B bij personen met een dubbele infectie met hiv-1 en HBV (zie de volledige voorschrijfinformatie voor EPIVIR-HBV). Het ontstaan van hepatitis B-virusvarianten die geassocieerd zijn met resistentie tegen lamivudine is ook gemeld bij met HIV-1 geïnfecteerde personen die lamivudine-bevattende antiretrovirale regimes hebben gekregen in aanwezigheid van een gelijktijdige infectie met het hepatitis B-virus.
Melkzuuracidose en ernstige hepatomegalie met steatose
Lactaatacidose en ernstige hepatomegalie met steatose, waaronder gevallen met fatale afloop, zijn gemeld bij het gebruik van nucleoside-analogen, waaronder EPIVIR. Een meerderheid van deze gevallen was bij vrouwen. Vrouwelijk geslacht en obesitas kunnen risicofactoren zijn voor de ontwikkeling van lactaatacidose en ernstige hepatomegalie met steatose bij patiënten die worden behandeld met antiretrovirale nucleoside-analogen. De behandeling met EPIVIR moet worden gestaakt bij elke patiënt die klinische of laboratoriumbevindingen ontwikkelt die wijzen op lactaatacidose of uitgesproken hepatotoxiciteit, waaronder hepatomegalie en steatose, zelfs bij afwezigheid van duidelijke transaminaseverhogingen.
Gebruik met op interferon en ribavirine gebaseerde regimes
In vitro-onderzoeken hebben aangetoond dat ribavirine de fosforylering van pyrimidine-nucleoside-analogen zoals lamivudine kan verminderen. Hoewel er geen bewijs van een farmacokinetische of farmacodynamische interactie (bijv. verlies van HIV-1/HCV-virologische onderdrukking) werd gezien wanneer ribavirine gelijktijdig werd toegediend met lamivudine bij patiënten met gelijktijdige HIV-1/HCV-infectie (zie KLINISCHE FARMACOLOGIE is leverdecompensatie (sommige fataal) opgetreden bij patiënten met een gelijktijdige infectie met hiv-1/HCV die een antiretrovirale combinatietherapie kregen voor hiv-1 en interferon alfa met of zonder ribavirine. Patiënten die interferon alfa met of zonder ribavirine en EPIVIR krijgen, moeten nauwlettend worden gecontroleerd op met de behandeling samenhangende toxiciteiten, met name leverdecompensatie. Stopzetting van EPIVIR 150 mg moet als medisch passend worden beschouwd. Dosisverlaging of stopzetting van interferon alfa, ribavirine of beide moet ook worden overwogen als verergering van klinische toxiciteiten wordt waargenomen, waaronder leverdecompensatie (bijv. Child-Pugh groter dan 6). Zie de volledige voorschrijfinformatie voor interferon en ribavirine.
Pancreatitis
Bij pediatrische patiënten met een voorgeschiedenis van eerdere blootstelling aan antiretrovirale nucleoside, een voorgeschiedenis van pancreatitis of andere significante risicofactoren voor de ontwikkeling van pancreatitis, dient EPIVIR 150 mg met voorzichtigheid te worden gebruikt. De behandeling met EPIVIR 150 mg moet onmiddellijk worden stopgezet als zich klinische tekenen, symptomen of laboratoriumafwijkingen voordoen die wijzen op pancreatitis [zie ONGEWENSTE REACTIES ].
Immuunreconstitutiesyndroom
Immuunreconstitutiesyndroom is gemeld bij patiënten die werden behandeld met antiretrovirale combinatietherapie, waaronder EPIVIR. Tijdens de beginfase van antiretrovirale combinatiebehandeling kunnen patiënten van wie het immuunsysteem reageert een ontstekingsreactie ontwikkelen op indolente of resterende opportunistische infecties (zoals Mycobacterium avium-infectie, cytomegalovirus, Pneumocystis jirovecii-pneumonie [PCP] of tuberculose), wat verdere evaluatie noodzakelijk kan maken en behandeling.
Van auto-immuunziekten (zoals de ziekte van Graves, polymyositis en Guillain-Barré-syndroom) is ook gemeld dat ze optreden in de setting van immuunreconstitutie, maar de tijd tot het begin is variabeler en kan vele maanden na het begin van de behandeling optreden.
Lagere virologische onderdrukkingspercentages en verhoogd risico op virale resistentie met orale oplossing
Pediatrische proefpersonen die op enig moment in het ARROW-onderzoek gelijktijdig met andere antiretrovirale orale oplossingen 150 mg EPIVIR drank (met op de gewichtsklasse gebaseerde doses van ongeveer 8 mg per kg per dag) kregen, hadden een lagere mate van virologische suppressie, een lagere plasmablootstelling aan lamivudine en ontwikkelden virale resistentie vaker dan degenen die EPIVIR-tabletten kregen [zie: KLINISCHE FARMACOLOGIE , Microbiologie , Klinische studies ].
EPIVIR-tablet met breukgleuf is de voorkeursformulering voor met HIV-1 geïnfecteerde pediatrische patiënten die ten minste 14 kg wegen en voor wie een vaste doseringsvorm geschikt is. Indien mogelijk moet een volledig tabletregime worden gebruikt om een mogelijke interactie met sorbitol te voorkomen [zie: KLINISCHE FARMACOLOGIE ]. Overweeg frequentere controle van de HIV-1 viral load bij behandeling met EPIVIR 150 mg drank.
Informatie over patiëntbegeleiding
Adviseer de patiënt om de door de FDA goedgekeurde patiëntetikettering (Patiëntinformatie) te lezen.
Patiënten met Hepatitis B of C Co-infectie
Informeer patiënten met gelijktijdige infectie met HIV-1 en HBV dat in sommige gevallen verslechtering van de leverziekte is opgetreden wanneer de behandeling met lamivudine werd stopgezet. Adviseer patiënten om eventuele veranderingen in het regime met hun zorgverlener te bespreken [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Informeer patiënten met gelijktijdige HIV-1/HCV-infectie dat leverdecompensatie (sommige fataal) is opgetreden bij patiënten met gelijktijdige HIV-1/HCV-infectie die antiretrovirale combinatietherapie voor HIV-1 en interferon alfa met of zonder ribavirine kregen [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Verschillen in formuleringen van EPIVIR
Adviseer patiënten dat EPIVIR 150 mg tabletten en drank een hogere dosis van hetzelfde werkzame bestanddeel (lamivudine) bevatten als EPIVIR-HBV tabletten en drank. Als wordt besloten om lamivudine op te nemen in het hiv-1-behandelingsschema van een patiënt die gelijktijdig is geïnfecteerd met hiv-1 en HBV, moeten de formulering en dosering van lamivudine in EPIVIR (niet EPIVIR-HBV) worden gebruikt [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Melkzuuracidose/hepatomegalie met steatose
Adviseer patiënten dat lactaatacidose en ernstige hepatomegalie met steatose zijn gemeld bij gebruik van nucleoside-analogen en andere antiretrovirale middelen. Adviseer patiënten om te stoppen met het gebruik van EPIVIR als ze klinische symptomen ontwikkelen die wijzen op lactaatacidose of uitgesproken hepatotoxiciteit [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Risico op pancreatitis
Adviseer ouders of voogden om pediatrische patiënten te controleren op tekenen en symptomen van pancreatitis [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Immuunreconstitutiesyndroom
Adviseer patiënten om hun zorgverlener onmiddellijk op de hoogte te stellen van eventuele tekenen en symptomen van infectie, aangezien een ontsteking door een eerdere infectie snel na antiretrovirale combinatietherapie kan optreden, ook wanneer EPIVIR wordt gestart [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Lagere virologische onderdrukkingspercentages en verhoogd risico op virale resistentie met orale oplossing
Adviseer patiënten dat indien mogelijk een volledig tabletregime moet worden gebruikt vanwege een verhoogde kans op falen van de behandeling bij pediatrische proefpersonen die EPIVIR drank gelijktijdig met andere antiretrovirale orale oplossingen kregen [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Sucrosegehalte van EPIVIR orale oplossing
Adviseer diabetespatiënten dat elke dosis van 15 ml EPIVIR drank 3 gram sucrose bevat (1 ml = 200 mg sucrose) (zie OMSCHRIJVING ].
Zwangerschapsregister
Adviseer patiënten dat er een zwangerschapsblootstellingsregister is dat de zwangerschapsuitkomsten controleert bij vrouwen die tijdens de zwangerschap aan EPIVIR zijn blootgesteld [zie Gebruik bij specifieke populaties ].
Borstvoeding
Instrueer vrouwen met een HIV-1-infectie om geen borstvoeding te geven, omdat HIV-1 via de moedermelk aan de baby kan worden doorgegeven [zie Gebruik bij specifieke populaties ].
Gemiste dosering
Instrueer patiënten dat ze, als ze een dosis EPIVIR 150 mg overslaan, deze moeten innemen zodra ze eraan denken. Adviseer patiënten om hun volgende dosis niet te verdubbelen of meer in te nemen dan de voorgeschreven dosis [zie: DOSERING EN ADMINISTRATIE ].
EPIVIR en RETROVIR zijn handelsmerken die eigendom zijn van of in licentie zijn gegeven aan de ViiV Healthcare-groep van bedrijven.
Niet-klinische toxicologie
Carcinogenese, mutagenese, verminderde vruchtbaarheid
Carcinogenese
Langdurige carcinogeniteitsstudies met lamivudine bij muizen en ratten toonden geen bewijs van carcinogeen potentieel bij blootstellingen tot 10 keer (muizen) en 58 keer (ratten) de menselijke blootstelling bij de aanbevolen dosis van 300 mg.
Mutagenese
Lamivudine was mutageen in een L5178Y muislymfoomtest en clastogeen in een cytogenetische test met behulp van gekweekte menselijke lymfocyten. Lamivudine was niet mutageen in een microbiële mutageniteitstest, in een in vitro celtransformatietest, in een micronucleustest bij ratten, in een beenmergcytogenetische test bij ratten en in een test voor ongeplande DNA-synthese in rattenlever. Lamivudine vertoonde geen bewijs van in vivo genotoxische activiteit bij ratten bij orale doses tot 2.000 mg per kg, waarbij plasmaspiegels werden geproduceerd van 35 tot 45 keer die bij mensen bij de aanbevolen dosis voor HIV-1-infectie.
Aantasting van de vruchtbaarheid
In een onderzoek naar reproductieve prestaties bleek dat lamivudine, toegediend aan ratten in doses tot 4.000 mg per kg per dag, plasmaspiegels produceerde die 47 tot 70 keer hoger waren dan die bij mensen, geen bewijs van verminderde vruchtbaarheid en geen effect op de overleving, groei en ontwikkeling aan het licht bracht. tot het spenen van het nageslacht.
Gebruik bij specifieke populaties
Zwangerschap
Zwangerschapsblootstellingsregister
Er is een register voor blootstelling aan zwangerschap dat de zwangerschapsuitkomsten controleert bij vrouwen die tijdens de zwangerschap aan EPIVIR zijn blootgesteld. Zorgaanbieders worden aangemoedigd om patiënten te registreren door het Antiretroviral Pregnancy Registry (APR) te bellen op 1-800-258-4263.
Risico Samenvatting
Beschikbare gegevens van het APR laten geen verschil zien in het algehele risico op geboorteafwijkingen voor lamivudine vergeleken met het achtergrondpercentage voor geboorteafwijkingen van 2,7% in de referentiepopulatie van het Metropolitan Atlanta Congenital Defects Program (MACDP) (zie Gegevens ). De APR gebruikt de MACDP als de Amerikaanse referentiepopulatie voor geboorteafwijkingen in de algemene bevolking. De MACDP evalueert vrouwen en zuigelingen uit een beperkt geografisch gebied en omvat geen uitkomsten voor geboorten die plaatsvonden bij een zwangerschapsduur van minder dan 20 weken. Het percentage miskraam wordt niet vermeld in het JKP. Het geschatte achtergrondpercentage van miskramen bij klinisch erkende zwangerschappen in de algemene bevolking van de VS is 15% tot 20%. Het achtergrondrisico op ernstige geboorteafwijkingen en miskramen voor de aangegeven populatie is niet bekend.
In reproductiestudies bij dieren resulteerde orale toediening van lamivudine aan drachtige konijnen tijdens de organogenese in embryoletaliteit bij systemische blootstelling (AUC) vergelijkbaar met de aanbevolen klinische dosis; er werden echter geen nadelige ontwikkelingseffecten waargenomen bij orale toediening van lamivudine aan drachtige ratten tijdens de organogenese bij plasmaconcentraties (Cmax) van 35 maal de aanbevolen klinische dosis (zie Gegevens ).
Gegevens
Menselijke gegevens
Op basis van prospectieve rapporten aan de APR van meer dan 11.000 blootstellingen aan lamivudine tijdens de zwangerschap resulterend in levendgeborenen (waaronder meer dan 4.500 blootgesteld in het eerste trimester), was er geen verschil tussen het algehele risico op geboorteafwijkingen voor lamivudine in vergelijking met het aantal geboorteafwijkingen op de achtergrond. van 2,7% in de Amerikaanse referentiepopulatie van het MACDP. De prevalentie van defecten bij levendgeborenen was 3,1% (95%-BI: 2,6% tot 3,6%) na blootstelling in het eerste trimester aan lamivudine-bevattende regimes en 2,8% (95%-BI: 2,5% tot 3,3%) na blootstelling in het tweede/derde trimester. aan lamivudine-bevattende regimes.
De farmacokinetiek van lamivudine is onderzocht bij zwangere vrouwen tijdens 2 klinische onderzoeken die in Zuid-Afrika zijn uitgevoerd. In de onderzoeken werd de farmacokinetiek beoordeeld bij 16 vrouwen bij 36 weken zwangerschap die 150 mg lamivudine tweemaal daags gebruikten met zidovudine, 10 vrouwen bij 38 weken zwangerschap die 150 mg lamivudine tweemaal daags gebruikten met zidovudine, en 10 vrouwen bij 38 weken zwangerschap die lamivudine 300 mg tweemaal gebruikten. dagelijks zonder andere antiretrovirale middelen. Deze onderzoeken waren niet ontworpen of ontwikkeld om informatie over de werkzaamheid te verschaffen. De lamivudineconcentraties waren over het algemeen vergelijkbaar in serummonsters van moeders, pasgeborenen en navelstrengen. Bij een subgroep van proefpersonen werden amnionvloeistofmonsters verzameld na natuurlijke breuk van de vliezen en bevestigden dat lamivudine de placenta passeert bij mensen. Op basis van beperkte gegevens bij de bevalling waren de mediane (spreiding) vruchtwaterconcentraties van lamivudine 3,9 (1,2 tot 12,8) maal hoger in vergelijking met gepaarde serumconcentraties van de moeder (n = 8).
Dierlijke gegevens
Lamivudine werd oraal toegediend aan drachtige ratten (met 90, 600 en 4.000 mg per kg per dag) en konijnen (met 90, 300 en 1.000 mg per kg per dag en met 15, 40 en 90 mg per kg per dag) tijdens de organogenese (op drachtdagen 7 tot 16 [rat] en 8 tot 20 [konijn]). Er zijn geen aanwijzingen gevonden voor foetale misvormingen als gevolg van lamivudine bij ratten en konijnen bij doses die plasmaconcentraties (Cmax) produceren die ongeveer 35 keer hoger zijn dan de blootstelling bij de mens bij de aanbevolen dagelijkse dosis. Bewijs van vroege embryonale letaliteit werd waargenomen bij konijnen bij systeemblootstellingen (AUC) vergelijkbaar met die waargenomen bij mensen, maar er waren geen aanwijzingen voor dit effect bij ratten bij plasmaconcentraties (Cmax) die 35 keer hoger waren dan de blootstelling bij de mens bij de aanbevolen dagelijkse dosis . Studies bij drachtige ratten hebben aangetoond dat lamivudine via de placenta op de foetus wordt overgedragen. In het vruchtbaarheids-/pre- en postnatale ontwikkelingsonderzoek bij ratten werd lamivudine oraal toegediend in doses van 180, 900 en 4.000 mg per kg per dag (van vóór de paring tot postnatale dag 20). In het onderzoek werd de ontwikkeling van het nageslacht, inclusief vruchtbaarheid en reproductieve prestaties, niet beïnvloed door toediening van lamivudine door de moeder.
Borstvoeding
Risico Samenvatting
De Centers for Disease Control and Prevention beveelt aan dat met HIV-1 geïnfecteerde moeders in de Verenigde Staten hun baby's geen borstvoeding geven om het risico van postnatale overdracht van HIV-1-infectie te vermijden. Lamivudine is aanwezig in moedermelk. Er is geen informatie over de effecten van lamivudine op de zuigeling die borstvoeding krijgt of de effecten van de geneesmiddelen op de melkproductie. Instrueer moeders om geen borstvoeding te geven vanwege de mogelijkheid van (1) HIV-1-overdracht (bij HIV-negatieve zuigelingen), (2) het ontwikkelen van virale resistentie (bij HIV-positieve zuigelingen) en (3) bijwerkingen bij een zuigeling die borstvoeding krijgt. als ze EPIVIR krijgen.
Pediatrisch gebruik
De veiligheid en werkzaamheid van EPIVIR 150 mg in combinatie met andere antiretrovirale middelen zijn vastgesteld bij pediatrische patiënten van 3 maanden en ouder. EPIVIR-tablet met breukgleuf is de voorkeursformulering voor met HIV-1 geïnfecteerde pediatrische patiënten die ten minste 14 kg wegen en voor wie een vaste doseringsvorm geschikt is omdat pediatrische proefpersonen die 150 mg EPIVIR drank kregen, een lagere virologische suppressie hadden en een lagere blootstelling aan plasmalamivudine. , en ontwikkelden vaker virale resistentie dan degenen die EPIVIR 150 mg-tabletten kregen in de ARROW-studie [zie DOSERING EN ADMINISTRATIE , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , ONGEWENSTE REACTIES , KLINISCHE FARMACOLOGIE , Klinische studies ].
Geriatrisch gebruik
Klinische onderzoeken met EPIVIR omvatten niet voldoende aantallen proefpersonen van 65 jaar en ouder om te bepalen of zij anders reageren dan jongere proefpersonen. Over het algemeen moet voorzichtigheid worden betracht bij de toediening van EPIVIR aan oudere patiënten vanwege de grotere frequentie van verminderde lever-, nier- of hartfunctie en van gelijktijdige ziekte of andere medicamenteuze behandeling [zie DOSERING EN ADMINISTRATIE , KLINISCHE FARMACOLOGIE ].
Patiënten met een verminderde nierfunctie
Verlaging van de dosering van EPIVIR 150 mg wordt aanbevolen voor patiënten met een verminderde nierfunctie [zie: DOSERING EN ADMINISTRATIE , KLINISCHE FARMACOLOGIE ].
OVERDOSERING
Er is geen specifieke behandeling bekend voor overdosering met EPIVIR. Als overdosering optreedt, moet de patiënt worden gecontroleerd en zo nodig een standaard ondersteunende behandeling worden toegepast. Omdat een verwaarloosbare hoeveelheid lamivudine werd verwijderd via (4-uurs) hemodialyse, continue ambulante peritoneale dialyse en geautomatiseerde peritoneale dialyse, is het niet bekend of continue hemodialyse klinisch voordeel zou opleveren bij een overdosis lamivudine.
CONTRA-INDICATIES
EPIVIR 150 mg is gecontra-indiceerd bij patiënten met een eerdere overgevoeligheidsreactie op lamivudine.
KLINISCHE FARMACOLOGIE
Werkingsmechanisme
Lamivudine is een antiretroviraal middel [zie Microbiologie ].
Farmacokinetiek
Farmacokinetiek bij volwassenen
De farmacokinetische eigenschappen van lamivudine zijn onderzocht bij asymptomatische, met HIV-1 geïnfecteerde volwassen proefpersonen na toediening van enkelvoudige intraveneuze (IV) doses variërend van 0,25 tot 8 mg per kg, evenals enkelvoudige en meervoudige (tweemaal daagse regime) orale doses variërend van 0,25 tot 10 mg per kg.
De farmacokinetische eigenschappen van lamivudine zijn ook onderzocht als enkelvoudige en meervoudige orale doses variërend van 5 mg tot 600 mg per dag toegediend aan met HBV geïnfecteerde personen.
De farmacokinetische eigenschappen bij steady-state van de EPIVIR 300 mg tablet eenmaal daags gedurende 7 dagen in vergelijking met de EPIVIR 150 mg tablet tweemaal daags gedurende 7 dagen werden beoordeeld in een cross-over studie bij 60 gezonde proefpersonen. EPIVIR 300 mg eenmaal daags resulteerde in blootstellingen aan lamivudine die vergelijkbaar waren met EPIVIR 150 mg tweemaal daags met betrekking tot plasma-AUC24,ss; de Cmax,ss was echter 66% hoger en de dalwaarde was 53% lager in vergelijking met het 150 mg tweemaal daags regime. De intracellulaire blootstelling aan lamivudinetrifosfaat in mononucleaire cellen in perifeer bloed was ook vergelijkbaar met betrekking tot AUC24,ss en Cmax24,ss; de dalwaarden waren echter lager in vergelijking met het schema van tweemaal daags 150 mg. De interindividuele variabiliteit was groter voor intracellulaire lamivudinetrifosfaatconcentraties versus lamivudineplasmadalconcentraties.

De farmacokinetiek van lamivudine werd geëvalueerd bij 12 volwassen met HIV-1 geïnfecteerde proefpersonen die tweemaal daags 150 mg lamivudine kregen toegediend in combinatie met andere antiretrovirale middelen. Het geometrische gemiddelde (95% BI) voor AUC(0-12) was 5,53 (4,58, 6,67) mcg.u per ml en voor Cmax was 1,40 (1,17, 1,69) mcg per ml.
Absorptie en biologische beschikbaarheid
De absolute biologische beschikbaarheid bij 12 volwassen proefpersonen was 86% ± 16% (gemiddelde ± SD) voor de 150 mg tablet en 87% ± 13% voor de drank. Na orale toediening van 2 mg per kg tweemaal daags aan 9 volwassenen met hiv-1, was de piekserumconcentratie van lamivudine (Cmax) 1,5 ± 0,5 mcg per ml (gemiddelde ± SD). De oppervlakte onder de plasmaconcentratie-versus-tijdcurve (AUC) en Cmax namen evenredig met de orale dosis toe binnen het bereik van 0,25 tot 10 mg per kg.
De accumulatieratio van lamivudine bij hiv-1-positieve asymptomatische volwassenen met een normale nierfunctie was 1,50 na 15 dagen orale toediening van 2 mg per kg tweemaal daags.
Effecten van voedsel op orale absorptie
EPIVIR 150 mg tabletten en drank kunnen met of zonder voedsel worden toegediend. Een experimentele doseringsvorm van 25 mg lamivudine werd tweemaal oraal toegediend aan 12 asymptomatische, met HIV-1 geïnfecteerde proefpersonen, eenmaal in nuchtere toestand en eenmaal met voedsel (1099 kcal; 75 gram vet, 34 gram eiwit, 72 gram koolhydraten ). De absorptie van lamivudine was langzamer in gevoede toestand (Tmax: 3,2 ± 1,3 uur) vergeleken met nuchtere toestand (Tmax: 0,9 ± 0,3 uur); Cmax in gevoede toestand was 40% ± 23% (gemiddelde ± SD) lager dan in nuchtere toestand. Er was geen significant verschil in systemische blootstelling (AUC∞) in gevoede en nuchtere toestand.
Verdeling
Het schijnbare distributievolume na intraveneuze toediening van lamivudine aan 20 proefpersonen was 1,3 ± 0,4 l per kg, wat erop wijst dat lamivudine zich in extravasculaire ruimten verspreidt. Het distributievolume was onafhankelijk van de dosis en correleerde niet met het lichaamsgewicht.
De binding van lamivudine aan humane plasma-eiwitten is minder dan 36%. In vitro-onderzoeken toonden aan dat over het concentratiebereik van 0,1 tot 100 mcg per ml, de hoeveelheid lamivudine geassocieerd met erytrocyten varieerde van 53% tot 57% en onafhankelijk was van de concentratie.
Metabolisme
Het metabolisme van lamivudine is een minder belangrijke eliminatieroute. Bij mensen is de enige bekende metaboliet van lamivudine de transsulfoxidemetaboliet (ongeveer 5% van een orale dosis na 12 uur). Serumconcentraties van deze metaboliet zijn niet bepaald. Lamivudine wordt niet significant gemetaboliseerd door cytochroom P450-enzymen.
Eliminatie
Het merendeel van lamivudine wordt onveranderd in de urine uitgescheiden door actieve organische kationische secretie. Bij 9 gezonde proefpersonen die een eenmalige orale dosis lamivudine van 300 mg kregen, was de renale klaring 199,7 ± 56,9 ml per minuut (gemiddelde ± SD). Bij 20 met HIV-1 geïnfecteerde proefpersonen die een enkele IV-dosis kregen, was de renale klaring 280,4 ± 75,2 ml per min (gemiddelde ± SD), wat neerkomt op 71% ± 16% (gemiddelde ± SD) van de totale klaring van lamivudine.
In de meeste onderzoeken met een enkelvoudige dosis bij met hiv-1 geïnfecteerde proefpersonen, met HBV geïnfecteerde proefpersonen of gezonde proefpersonen met serummonsters gedurende 24 uur na toediening, varieerde de waargenomen gemiddelde eliminatiehalfwaardetijd (t½) van 5 tot 7 uur. Bij met HIV-1 geïnfecteerde proefpersonen was de totale klaring 398,5 ± 69,1 ml per minuut (gemiddelde ± SD). De orale klaring en eliminatiehalfwaardetijd waren onafhankelijk van de dosis en het lichaamsgewicht over een oraal doseringsbereik van 0,25 tot 10 mg per kg.
Specifieke populaties
Patiënten met nierinsufficiëntie
De farmacokinetische eigenschappen van lamivudine zijn vastgesteld bij een kleine groep met HIV-1 geïnfecteerde volwassenen met een verminderde nierfunctie (Tabel 7).
Tmax werd niet significant beïnvloed door de nierfunctie. Op basis van deze waarnemingen wordt aanbevolen de dosering van lamivudine aan te passen bij patiënten met nierinsufficiëntie [zie: DOSERING EN ADMINISTRATIE ].
Op basis van een onderzoek bij overigens gezonde proefpersonen met een verminderde nierfunctie verhoogde hemodialyse de klaring van lamivudine van gemiddeld 64 tot 88 ml per minuut; de duur van hemodialyse (4 uur) was echter onvoldoende om de gemiddelde blootstelling aan lamivudine significant te veranderen na toediening van een enkelvoudige dosis. Continue ambulante peritoneale dialyse en geautomatiseerde peritoneale dialyse hebben verwaarloosbare effecten op de klaring van lamivudine. Daarom wordt aanbevolen om, na correctie van de dosis voor creatinineklaring, geen aanvullende dosisaanpassingen te maken na routinematige hemodialyse of peritoneale dialyse.
De effecten van nierinsufficiëntie op de farmacokinetiek van lamivudine bij pediatrische patiënten zijn niet bekend.
Patiënten met leverinsufficiëntie
De farmacokinetische eigenschappen van lamivudine zijn vastgesteld bij volwassenen met een verminderde leverfunctie. Farmacokinetische parameters werden niet veranderd door een verminderde leverfunctie. De veiligheid en werkzaamheid van lamivudine zijn niet vastgesteld in de aanwezigheid van gedecompenseerde leverziekte.
Zwangere vrouw
De farmacokinetiek van lamivudine is onderzocht bij 36 zwangere vrouwen tijdens 2 klinische onderzoeken in Zuid-Afrika. De farmacokinetiek van lamivudine bij zwangere vrouwen was vergelijkbaar met die bij niet-zwangere volwassenen en bij postpartum vrouwen. De lamivudineconcentraties waren over het algemeen vergelijkbaar in serummonsters van moeders, pasgeborenen en navelstrengen.
Pediatrische patiënten
De farmacokinetiek van lamivudine is onderzocht na eenmalige of herhaalde doses EPIVIR bij 210 pediatrische proefpersonen. Pediatrische patiënten die lamivudine drank kregen (gedoseerd met ongeveer 8 mg per kg per dag) bereikten ongeveer 25% lagere plasmaconcentraties van lamivudine in vergelijking met met HIV-1 geïnfecteerde volwassenen. Pediatrische patiënten die lamivudine orale tabletten kregen, bereikten plasmaconcentraties die vergelijkbaar waren met of iets hoger waren dan die waargenomen bij volwassenen. De absolute biologische beschikbaarheid van zowel EPIVIR-tabletten als drank is lager bij kinderen dan bij volwassenen. De relatieve biologische beschikbaarheid van EPIVIR drank is ongeveer 40% lager dan die van tabletten die lamivudine bevatten bij pediatrische patiënten, ondanks dat er geen verschil is bij volwassenen. Lagere blootstelling aan lamivudine bij pediatrische patiënten die EPIVIR 150 mg drank krijgen, is waarschijnlijk te wijten aan de interactie tussen lamivudine en gelijktijdige oplossingen die sorbitol bevatten (zoals ZIAGEN). Modellering van farmacokinetische gegevens suggereert dat verhoging van de dosering van EPIVIR drank tot 5 mg per kg oraal tweemaal daags of 10 mg per kg oraal eenmaal daags (tot een maximum van 300 mg per dag) nodig is om voldoende concentraties van lamivudine te bereiken [zie DOSERING EN ADMINISTRATIE ]. Er zijn geen klinische gegevens bij met HIV-1 geïnfecteerde pediatrische patiënten die in deze dosis gelijktijdig werden toegediend met sorbitolbevattende geneesmiddelen.
De farmacokinetiek van eenmaal daags gedoseerde lamivudine bij met hiv-1 geïnfecteerde pediatrische proefpersonen in de leeftijd van 3 maanden tot 12 jaar werd geëvalueerd in 3 onderzoeken (PENTA-15 [n = 17], PENTA 13 [n = 19] en ARROW PK [n = 35]). Alle 3 de onderzoeken waren cross-over, open-label farmacokinetische onderzoeken met 2 perioden van tweemaal daagse dosering van abacavir en lamivudine. Deze 3 onderzoeken toonden aan dat een eenmaaldaagse dosering een vergelijkbare AUC0-24 geeft als een tweemaaldaagse dosering van lamivudine bij dezelfde totale dagelijkse dosis wanneer de doseringsschema's binnen dezelfde formulering worden vergeleken (dwz ofwel de drank ofwel de tabletformulering). De gemiddelde Cmax was ongeveer 80% tot 90% hoger bij een eenmaaldaagse dosering van lamivudine dan bij een tweemaaldaagse dosering.
Distributie van lamivudine in cerebrospinale vloeistof (CSF) werd beoordeeld bij 38 pediatrische proefpersonen na meervoudige orale toediening van lamivudine. CSF-monsters werden tussen 2 en 4 uur na de dosis verzameld. Bij een dosis van 8 mg per kg per dag varieerden de CSF-lamivudineconcentraties bij 8 proefpersonen van 5,6% tot 30,9% (gemiddelde ± SD van 14,2% ± 7,9%) van de concentratie in een gelijktijdig serummonster, met CSF-lamivudineconcentraties variërend van 0,04 tot 0,3 mcg per ml.
Er zijn beperkte, ongecontroleerde farmacokinetische en veiligheidsgegevens beschikbaar over de toediening van lamivudine (en zidovudine) aan 36 zuigelingen tot 1 week in 2 onderzoeken in Zuid-Afrika. In deze onderzoeken was de klaring van lamivudine aanzienlijk verminderd bij pasgeborenen van 1 week oud in vergelijking met eerder onderzochte pediatrische proefpersonen (ouder dan 3 maanden). Er is onvoldoende informatie om het tijdsverloop van veranderingen in de klaring vast te stellen tussen de onmiddellijke neonatale periode en de leeftijdsgroepen ouder dan 3 maanden [zie ONGEWENSTE REACTIES ].
Geriatrische patiënten
De farmacokinetiek van lamivudine na toediening van EPIVIR 150 mg aan personen ouder dan 65 jaar is niet onderzocht [zie Gebruik bij specifieke populaties ].
Mannelijke en vrouwelijke patiënten
Er zijn geen significante of klinisch relevante sekseverschillen in de farmacokinetiek van lamivudine.
Raciale groepen
Er zijn geen significante of klinisch relevante raciale verschillen in de farmacokinetiek van lamivudine.
Geneesmiddelinteractiestudies
Effect van lamivudine op de farmacokinetiek van andere middelen
Op basis van in vitro onderzoeksresultaten wordt niet verwacht dat lamivudine bij blootstelling aan therapeutische geneesmiddelen de farmacokinetiek beïnvloedt van geneesmiddelen die substraten zijn van de volgende transporteiwitten: organisch aniontransporteiwit 1B1/3 (OATP1B1/3), borstkankerresistentie-eiwit (BCRP), P-glycoproteïne (P-gp), multidrug en toxine-extrusie-eiwit 1 (MATE)1, MATE2-K, organische kationtransporter 1 (OCT)1, OCT2 of OCT3.
Effect van andere middelen op de farmacokinetiek van lamivudine
Lamivudine is in vitro een substraat van MATE1, MATE2-K en OCT2. Van trimethoprim (een remmer van deze geneesmiddeltransporters) is aangetoond dat het de plasmaconcentraties van lamivudine verhoogt. Deze interactie wordt niet als klinisch significant beschouwd aangezien er geen dosisaanpassing van lamivudine nodig is.
Lamivudine is een substraat van P-gp en BCRP; gezien de absolute biologische beschikbaarheid (87%), is het echter onwaarschijnlijk dat deze transporters een significante rol spelen bij de absorptie van lamivudine. Daarom is het onwaarschijnlijk dat gelijktijdige toediening van geneesmiddelen die deze effluxtransporters remmen, de dispositie en eliminatie van lamivudine beïnvloedt.
Interferon Alfa
Er was geen significante farmacokinetische interactie tussen lamivudine en interferon alfa in een onderzoek met 19 gezonde mannelijke proefpersonen [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Ribavirine
In vitro-gegevens wijzen erop dat ribavirine de fosforylering van lamivudine, stavudine en zidovudine vermindert. Er werd echter geen farmacokinetische (bijv. plasmaconcentraties of intracellulaire trifosforyleerde concentraties van actieve metabolieten) of farmacodynamische (bijv. verlies van HIV-1/HCV-virologische onderdrukking) waargenomen wanneer ribavirine en lamivudine (n = 18), stavudine (n = 10) , of zidovudine (n = 6) werden gelijktijdig toegediend als onderdeel van een multidrugregime aan personen met een gelijktijdige HIV-1/HCV-infectie [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Sorbitol (Excipiëns)
Lamivudine- en sorbitoloplossingen werden gelijktijdig toegediend aan 16 gezonde volwassen proefpersonen in een open-label, gerandomiseerde, 4-periode cross-over studie. Elke proefpersoon kreeg een enkele dosis van 300 mg lamivudine drank alleen of gelijktijdig toegediend met een enkele dosis van 3,2 gram, 10,2 gram of 13,4 gram sorbitol in oplossing. Gelijktijdige toediening van lamivudine met sorbitol resulteerde in dosisafhankelijke verlagingen van 20%, 39% en 44% in de AUC(0-24), 14%, 32% en 36% in de AUC(∞) en 28%, 52% en 55% in de Cmax; respectievelijk lamivudine.
Trimethoprim/Sulfamethoxazol
Lamivudine en TMP/SMX werden gelijktijdig toegediend aan 14 hiv-1-positieve proefpersonen in een single-center, open-label, gerandomiseerde, cross-over studie. Elke proefpersoon kreeg een behandeling met een enkele dosis van 300 mg lamivudine en TMP 160 mg/SMX 800 mg eenmaal daags gedurende 5 dagen met gelijktijdige toediening van 300 mg lamivudine met de vijfde dosis in een gekruiste opzet. Gelijktijdige toediening van TMP/SMX met lamivudine resulteerde in een toename van 43% ± 23% (gemiddelde ± SD) in de AUC∞ van lamivudine, een afname van 29% ± 13% in de orale klaring van lamivudine en een afname van 30% ± 36% in lamivudine renale klaring. De farmacokinetische eigenschappen van TMP en SMX werden niet veranderd door gelijktijdige toediening met lamivudine. Er is geen informatie over het effect op de farmacokinetiek van lamivudine van hogere doses TMP/SMX zoals die worden gebruikt bij de behandeling van PCP.
Zidovudine
Er werden geen klinisch significante veranderingen in de farmacokinetiek van lamivudine of zidovudine waargenomen bij 12 asymptomatische met HIV-1 geïnfecteerde volwassen proefpersonen die een enkelvoudige dosis zidovudine (200 mg) kregen in combinatie met meerdere doses lamivudine (300 mg elke 12 uur).
Microbiologie
Werkingsmechanisme
Lamivudine is een synthetisch nucleoside-analoog. Intracellulair wordt lamivudine gefosforyleerd tot zijn actieve 5'-trifosfaatmetaboliet, lamivudinetrifosfaat (3TC-TP). Het belangrijkste werkingsmechanisme van 3TC-TP is remming van HIV-1 reverse transcriptase (RT) via DNA-ketenbeëindiging na opname van de nucleotide-analoog.
Antivirale activiteit
De antivirale activiteit van lamivudine tegen HIV-1 werd beoordeeld in een aantal cellijnen, waaronder monocyten en verse humane perifere bloedlymfocyten (PBMC's) met behulp van standaard gevoeligheidstesten. EC50-waarden lagen in het bereik van 0,003 tot 15 microM (1 microM = 0,23 mcg per ml). De mediane EC50-waarden van lamivudine waren 60 nM (bereik: 20 tot 70 nM), 35 nM (bereik: 30 tot 40 nM), 30 nM (bereik: 20 tot 90 nM), 20 nM (bereik: 3 tot 40 nM) , 30 nM (bereik: 1 tot 60 nM), 30 nM (bereik: 20 tot 70 nM), 30 nM (bereik: 3 tot 70 nM) en 30 nM (bereik: 20 tot 90 nM) tegen HIV-1-clades AG- en groep O-virussen (n = 3 behalve n = 2 voor clade B) respectievelijk. De EC50-waarden tegen HIV-2-isolaten (n = 4) varieerden van 0,003 tot 0,120 microM in PBMC's. Lamivudine was niet antagonistisch voor alle geteste anti-hiv-middelen. Ribavirine (50 microM), gebruikt bij de behandeling van chronische HCV-infectie, verminderde de anti-HIV-1-activiteit van lamivudine met een factor 3,5 in MT-4-cellen.
Weerstand
Lamivudine-resistente varianten van HIV-1 zijn geselecteerd in celkweek. Genotypische analyse toonde aan dat de resistentie het gevolg was van een specifieke aminozuursubstitutie in het HIV-1 reverse transcriptase op codon 184 waarbij de methionine veranderde in valine of isoleucine (M184V/I).
HIV-1-stammen die resistent zijn tegen zowel lamivudine als zidovudine zijn geïsoleerd uit proefpersonen. De gevoeligheid van klinische isolaten voor lamivudine en zidovudine werd gecontroleerd in gecontroleerde klinische onderzoeken. Bij proefpersonen die lamivudine monotherapie of combinatietherapie met lamivudine plus zidovudine kregen, werden hiv-1-isolaten van de meeste proefpersonen binnen 12 weken fenotypisch en genotypisch resistent tegen lamivudine.
Genotypische en fenotypische analyse van on-therapie hiv-1 isolaten van proefpersonen met virologisch falen
Proef EPV20001
Drieënvijftig van de 554 (10%) proefpersonen die deelnamen aan EPV20001 werden geïdentificeerd als virologisch falen (plasma hiv-1 RNA-niveau hoger dan of gelijk aan 400 kopieën per ml) in week 48. Achtentwintig proefpersonen werden gerandomiseerd naar de lamivudine eenmaal- dagelijkse behandelingsgroep en 25 tot de tweemaal daagse behandelingsgroep met lamivudine. De mediane baseline plasma hiv-1 RNA-spiegels van proefpersonen in de lamivudine eenmaal daags groep en lamivudine tweemaal daags groep waren respectievelijk 4,9 log10 kopieën per ml en 4,6 log10 kopieën per ml.
Genotypische analyse van isolaten tijdens behandeling van 22 proefpersonen geïdentificeerd als virologisch falen in de lamivudine-groep die eenmaal daags was, toonde aan dat isolaten van 8 van de 22 proefpersonen een tijdens de behandeling optredende substitutie met lamivudineresistentie (M184V of M184I) bevatten, isolaten van 0 van 22 proefpersonen bevatten tijdens de behandeling optredende aminozuursubstituties geassocieerd met zidovudine-resistentie (M41L, D67N, K70R, L210W, T215Y/F of K219Q/E), en isolaten van 10 van de 22 proefpersonen bevatten tijdens de behandeling optredende aminozuursubstituties geassocieerd met efavirenz-resistentie ( L100I, K101E, K103N, V108I of Y181C).
Genotypische analyse van isolaten die tijdens de behandeling werden behandeld van proefpersonen (n = 22) in de groep die tweemaal daags met lamivudine werd behandeld, toonde aan dat isolaten van 5 van de 22 proefpersonen tijdens de behandeling optredende substituties voor lamivudine-resistentie bevatten, isolaten van 1 van de 22 proefpersonen vertoonden tijdens de behandeling optredende zidovudine-resistentie substituties en isolaten van 7 van de 22 proefpersonen bevatten tijdens de behandeling optredende efavirenz-resistentiesubstituties.
Fenotypische analyse van hiv-1-isolaten die op baseline overeenkomen met hiv-1-isolaten van proefpersonen (n=13) die eenmaal daags lamivudine kregen, toonde aan dat isolaten van 7 van de 13 proefpersonen een 85 tot 299-voudige afname in gevoeligheid voor lamivudine vertoonden, isolaten van 12 van 13 proefpersonen waren gevoelig voor zidovudine en isolaten van 8 van de 13 proefpersonen vertoonden een 25 tot 295-voudige afname in gevoeligheid voor efavirenz.
Fenotypische analyse van op baseline gematchte hiv-1-isolaten tijdens behandeling van proefpersonen (n = 13) die tweemaal daags lamivudine kregen, toonde aan dat isolaten van 4 van de 13 proefpersonen een 29- tot 159-voudige afname in gevoeligheid voor lamivudine vertoonden, isolaten van alle 13 proefpersonen waren gevoelig voor zidovudine en isolaten van 3 van de 13 proefpersonen vertoonden een 21- tot 342-voudige afname in gevoeligheid voor efavirenz.
Proef EPV40001
Vijftig proefpersonen kregen lamivudine 300 mg eenmaal daags plus zidovudine 300 mg tweemaal daags plus abacavir 300 mg tweemaal daags en 50 proefpersonen kregen lamivudine 150 mg plus zidovudine 300 mg plus abacavir 300 mg tweemaal daags. De mediane baseline plasma hiv-1 RNA-spiegels voor proefpersonen in de 2 groepen waren respectievelijk 4,79 log10 kopieën per ml en 4,83 log10 kopieën per ml. Veertien van de 50 proefpersonen in de groep die eenmaal daags met lamivudine werd behandeld en 9 van de 50 proefpersonen in de groep die tweemaal daags met lamivudine werd behandeld, werden geïdentificeerd als virologisch falen.
Genotypische analyse van hiv-1-isolaten tijdens behandeling van proefpersonen (n = 9) in de groep die eenmaal daags met lamivudine werd behandeld, toonde aan dat isolaten van 6 proefpersonen een abacavir- en/of lamivudine-resistentie-geassocieerde substitutie M184V alleen hadden. Isolaten tijdens behandeling van proefpersonen (n = 6) die tweemaal daags lamivudine kregen, toonden aan dat isolaten van 2 proefpersonen alleen M184V hadden, en isolaten van 2 proefpersonen vertoonden de M184V-substitutie in combinatie met zidovudine-resistentie-geassocieerde aminozuursubstituties.
Fenotypische analyse van isolaten die onder behandeling waren van proefpersonen (n = 6) die eenmaal daags lamivudine kregen, toonde aan dat hiv-1-isolaten van 4 proefpersonen een 32- tot 53-voudige afname in gevoeligheid voor lamivudine vertoonden. Hiv-1-isolaten van deze 6 personen waren gevoelig voor zidovudine.
Fenotypische analyse van isolaten die onder behandeling waren van proefpersonen (n = 4) die tweemaal daags lamivudine kregen, toonde aan dat hiv-1-isolaten van 1 proefpersoon een 45-voudige afname in gevoeligheid voor lamivudine en een 4,5-voudige afname in gevoeligheid voor zidovudine vertoonden.
Kindergeneeskunde
Pediatrische patiënten die lamivudine drank gelijktijdig met andere antiretrovirale orale oplossingen (abacavir, nevirapine/efavirenz of zidovudine) kregen in ARROW, ontwikkelden vaker virale resistentie dan degenen die tabletten kregen. Bij randomisatie naar een eenmaal daagse of tweemaal daagse dosering van EPIVIR plus abacavir had 13% van de proefpersonen die begonnen met tabletten en 32% van de proefpersonen die begonnen met de oplossing resistentiesubstituties. Het bij pediatrische patiënten waargenomen resistentieprofiel is vergelijkbaar met het bij volwassenen waargenomen resistentieprofiel wat betreft de gedetecteerde genotypische substituties en relatieve frequentie, met de meest waargenomen substituties op M184 (V of I) [zie Klinische studies ].
Kruisweerstand
Er is kruisresistentie waargenomen bij nucleoside reverse transcriptase-remmers (NRTI's). Lamivudine-resistente HIV-1-mutanten waren kruisresistent in celkweek tegen didanosine (ddI). Kruisresistentie wordt ook verwacht met abacavir en emtricitabine, aangezien deze M184V-substituties selecteren.
Klinische studies
Het gebruik van EPIVIR is gebaseerd op de resultaten van klinische onderzoeken bij met hiv-1 geïnfecteerde proefpersonen in combinatieregimes met andere antiretrovirale middelen. Informatie uit onderzoeken met klinische eindpunten of een combinatie van CD4+-celtellingen en HIV-1-RNA-metingen is hieronder opgenomen als documentatie van de bijdrage van lamivudine aan een combinatieregime in gecontroleerde onderzoeken.
Volwassen onderwerpen
Klinische eindpuntproef
NUCB3007 (CAESAR) was een dubbelblind, placebogecontroleerd multicenteronderzoek waarin de voortzetting van de huidige therapie (alleen zidovudine [62% van de proefpersonen] of zidovudine met didanosine of zalcitabine [38% van de proefpersonen]) werd vergeleken met de toevoeging van EPIVIR 150 mg of EPIVIR 150 mg plus een niet-nucleoside reverse-transcriptaseremmer (NNRTI) in onderzoek, gerandomiseerd 1:2:1. Een totaal van 1.816 met hiv-1 geïnfecteerde volwassenen met 25 tot 250 CD4+-cellen per mm³ (mediaan = 122 cellen per mm³) bij aanvang werden geïncludeerd: de mediane leeftijd was 36 jaar, 87% was man, 84% had ervaring met nucleoside, en 16% was therapienaïef. De mediane duur van de proef was 12 maanden. De resultaten zijn samengevat in Tabel 9.
Surrogate Endpoint Trials
Dubbele nucleoside analoge proeven
In de belangrijkste klinische onderzoeken naar de initiële ontwikkeling van lamivudine werden combinaties van lamivudine/zidovudine vergeleken met zidovudine monotherapie of met zidovudine plus zalcitabine. Deze onderzoeken toonden het antivirale effect van lamivudine in een combinatie van twee geneesmiddelen aan. Meer recente toepassingen van lamivudine bij de behandeling van HIV-1-infectie omvatten het in meervoudige geneesmiddelenregimes die ten minste 3 antiretrovirale geneesmiddelen bevatten voor verbeterde virale onderdrukking.
Vergelijking van doseringsschema's Surrogaat-eindpuntonderzoeken bij therapienaïeve volwassenen
EPV20001 was een multicenter, dubbelblinde, gecontroleerde studie waarin proefpersonen 1:1 werden gerandomiseerd om EPIVIR 300 mg eenmaal daags of EPIVIR 150 mg tweemaal daags te krijgen, in combinatie met zidovudine 300 mg tweemaal daags en efavirenz 600 mg eenmaal daags. Een totaal van 554 antiretrovirale behandelingsnaïeve hiv-1-geïnfecteerde volwassenen namen deel: man (79%), blank (50%), mediane leeftijd van 35 jaar, baseline CD4+-celtellingen van 69 tot 1.089 cellen per mm³ (mediaan = 362 cellen per mm³) en mediane baseline plasma hiv-1 RNA van 4,66 log10 kopieën per ml. De resultaten van de behandeling tot 48 weken zijn samengevat in figuur 1 en tabel 10.
Afbeelding 1: Virologische respons tot en met week 48, EPV20001a,b (intent-to-treat) 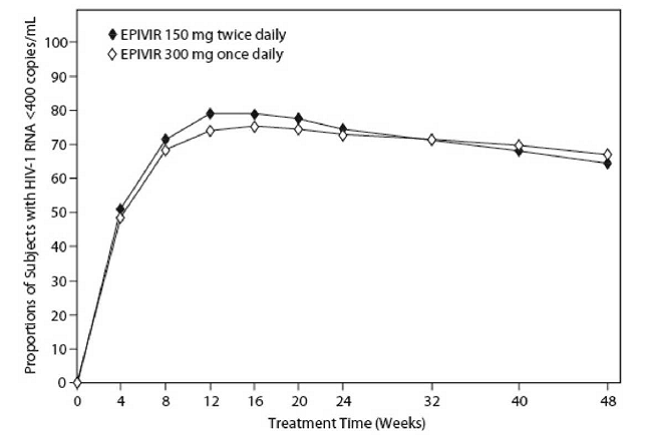
een Roche AMPLICOR HIV-1-MONITOR. b Responders bij elk bezoek zijn proefpersonen die bij dat bezoek minder dan 400 kopieën per ml hiv-1-RNA hadden bereikt en behouden zonder te stoppen.
Het aandeel proefpersonen met hiv-1-RNA van minder dan 50 kopieën per ml (via Roche Ultrasensitive assay) tot en met week 48 was 61% voor proefpersonen die eenmaal daags 300 mg EPIVIR kregen en 63% voor proefpersonen die tweemaal daags 150 mg EPIVIR kregen. De mediane toename van het aantal CD4+-cellen was 144 cellen per mm³ in week 48 bij proefpersonen die eenmaal daags 300 mg EPIVIR kregen en 146 cellen per mm³ bij proefpersonen die tweemaal daags 150 mg EPIVIR kregen.
Een kleine, gerandomiseerde, open-label proefstudie, EPV40001, werd uitgevoerd in Thailand. In totaal werden 159 behandelingsnaïeve volwassen proefpersonen (mannelijk 32%, Aziatisch 100%, mediane leeftijd 30 jaar, mediane CD4+-celtelling bij baseline 380 cellen per mm³, mediane plasma hiv-1 RNA 4,8 log10 kopieën per ml) geïncludeerd. Twee van de behandelarmen in dit onderzoek gaven een vergelijking tussen lamivudine 300 mg eenmaal daags (n = 54) en lamivudine 150 mg tweemaal daags (n = 52), elk in combinatie met zidovudine 300 mg tweemaal daags en abacavir 300 mg tweemaal daags. In intent-to-treat-analyses van gegevens van 48 weken was het aandeel proefpersonen met hiv-1-RNA van minder dan 400 kopieën per ml 61% (33 van de 54) in de groep die was gerandomiseerd naar eenmaal daags lamivudine en 75% (39 van de 52) in de groep die gerandomiseerd was om alle 3 de geneesmiddelen tweemaal daags te ontvangen; de verhoudingen met hiv-1-RNA van minder dan 50 kopieën per ml waren 54% (29 van de 54) in de eenmaal daagse lamivudine-groep en 67% (35 van de 52) in de alle-tweemaal-daagse groep; en de mediane toename van het aantal CD4+-cellen was 166 cellen per mm³ in de eenmaal daagse lamivudine-groep en 216 cellen per mm³ in de alle-tweemaal daagse groep.
Pediatrische onderwerpen
Klinische eindpuntproef
ACTG300 was een multicenter, gerandomiseerde, dubbelblinde studie waarin EPIVIR plus RETROVIR (zidovudine) werd vergeleken met didanosine-monotherapie. Een totaal van 471 symptomatische, met hiv-1 geïnfecteerde therapie-naïeve (minder dan of gelijk aan 56 dagen antiretrovirale therapie) pediatrische proefpersonen werden opgenomen in deze 2 behandelingsarmen. De mediane leeftijd was 2,7 jaar (bereik: 6 weken tot 14 jaar), 58% was vrouw en 86% was niet-blank. Het gemiddelde aantal CD4+-cellen bij baseline was 868 cellen per mm³ (gemiddeld: 1.060 cellen per mm³ en bereik: 0 tot 4.650 cellen per mm³ voor proefpersonen jonger dan of gelijk aan 5 jaar; gemiddeld: 419 cellen per mm³ en bereik: 0 tot 1.555 cellen per mm³ voor proefpersonen ouder dan 5 jaar) en de gemiddelde baseline plasma hiv-1 RNA was 5,0 log10 kopieën per ml. De mediane duur van het onderzoek was 10,1 maanden voor de proefpersonen die EPIVIR 150 mg plus RETROVIR kregen en 9,2 maanden voor de proefpersonen die monotherapie met didanosine kregen. De resultaten zijn samengevat in Tabel 11.
Eenmaal daagse dosering
ARROW (COL105677) was een 5 jaar durende, gerandomiseerde, multicenter studie waarin meerdere aspecten van de klinische behandeling van hiv-1-infectie bij pediatrische proefpersonen werden geëvalueerd. Met hiv-1 geïnfecteerde, niet eerder behandelde proefpersonen in de leeftijd van 3 maanden tot 17 jaar werden geïncludeerd en behandeld met een eerstelijnsbehandeling met EPIVIR 150 mg en abacavir, tweemaal daags gedoseerd volgens de aanbevelingen van de Wereldgezondheidsorganisatie. Na een behandeling van minimaal 36 weken kregen proefpersonen de mogelijkheid om deel te nemen aan Randomisatie 3 van het ARROW-onderzoek, waarbij de veiligheid en werkzaamheid van een eenmaal daagse dosering werd vergeleken met een tweemaal daagse dosering van EPIVIR en abacavir, in combinatie met een derde antiretroviraal middel. medicijn, voor nog eens 96 weken. Van de 1206 oorspronkelijke ARROW-proefpersonen namen 669 deel aan Randomisatie 3. Virologische suppressie was geen vereiste voor deelname: bij aanvang van Randomisatie 3 (na minimaal 36 weken tweemaal daagse behandeling), 75% van de proefpersonen in de tweemaal daagse cohort waren virologisch onderdrukt, vergeleken met 71% van de proefpersonen in het eenmaal daagse cohort.
Het aandeel proefpersonen met hiv-1-RNA van minder dan 80 kopieën per ml gedurende 96 weken wordt weergegeven in tabel 12. De verschillen tussen de virologische responsen in de twee behandelingsarmen waren vergelijkbaar over de baselinekenmerken voor geslacht en leeftijd.
Analyses per formulering toonden aan dat het aandeel proefpersonen met hiv-1-RNA van minder dan 80 kopieën per ml bij randomisatie en week 96 hoger was bij proefpersonen die tabletformuleringen van EPIVIR 150 mg en abacavir hadden gekregen (75% [458/610] en 72% [434/601]) dan bij degenen die oplossingsformulering(en) hadden gekregen (met EPIVIR 150 mg oplossing gegeven in op gewichtsband gebaseerde doses van ongeveer 8 mg per kg per dag) op enig moment (52% [29/56] en 54 % [30/56]), respectievelijk [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. Deze verschillen werden waargenomen in elke verschillende geëvalueerde leeftijdsgroep.
PATIËNT INFORMATIE
EPIVIR (EP-i-veer) (lamivudine) tabletten
EPIVIR (EP-i-veer) (lamivudine) drank
Wat is de belangrijkste informatie die ik moet weten over EPIVIR 150 mg?
EPIVIR kan ernstige bijwerkingen veroorzaken, waaronder:
- Verergering van het hepatitis B-virus bij mensen met een hiv-1-infectie. Als u hiv-1 (humaan immunodeficiëntievirus type 1) en hepatitis B-virus (HBV) heeft, kan uw HBV verergeren (opflakkering) als u stopt met het gebruik van EPIVIR. Een "flare-up" is wanneer uw HBV-infectie plotseling, erger dan voorheen, terugkeert. Een verslechtering van de leverziekte kan ernstig zijn en tot de dood leiden.
- Laat EPIVIR niet opraken. Vul uw recept bij of praat met uw zorgverlener voordat uw EPIVIR helemaal op is.
- Stop niet met EPIVIR zonder eerst met uw zorgverlener te overleggen.
- Als u stopt met het innemen van EPIVIR, zal uw zorgverlener uw gezondheid vaak moeten controleren en gedurende enkele maanden regelmatig bloedonderzoek moeten doen om uw lever te controleren.
- Resistent hepatitis B-virus (HBV). Als u hiv-1 en hepatitis B heeft, kan het hepatitis B-virus tijdens uw behandeling met EPIVIR 150 mg veranderen (muteren) en moeilijker te behandelen (resistent) worden.
- Gebruik met op interferon en ribavirine gebaseerde regimes. Verergering van een leverziekte die de dood heeft veroorzaakt, is opgetreden bij mensen die besmet zijn met zowel het hiv-1- als het hepatitis C-virus die antiretrovirale geneesmiddelen gebruiken en die ook worden behandeld voor hepatitis C met interferon met of zonder ribavirine. Als u EPIVIR 150 mg en interferon met of zonder ribavirine gebruikt, vertel het uw zorgverlener dan als u nieuwe symptomen heeft.
Wat is EPIVIR?
EPIVIR is een receptgeneesmiddel dat samen met andere antiretrovirale geneesmiddelen wordt gebruikt voor de behandeling van een infectie met het humaan immunodeficiëntievirus (hiv-1).
HIV-1 is het virus dat Acquired Immune Deficiency Syndrome (AIDS) veroorzaakt.
EPIVIR-tabletten en -drank (gebruikt voor de behandeling van HIV-1-infectie) bevatten een hogere dosis van hetzelfde werkzame bestanddeel (lamivudine) dan in het geneesmiddel EPIVIR-HBV-tabletten en -drank (gebruikt voor de behandeling van HBV). Als u zowel hiv-1 als HBV heeft, mag u EPIVIR-HBV niet gebruiken om uw infecties te behandelen.
De veiligheid en werkzaamheid van EPIVIR zijn niet vastgesteld bij kinderen jonger dan 3 maanden.
Wie mag EPIVIR 150 mg niet gebruiken?
Neem EPIVIR . niet in als u allergisch bent voor lamivudine of voor één van de bestanddelen van EPIVIR. Zie het einde van deze patiëntenbijsluiter voor een volledige lijst van ingrediënten in EPIVIR.
Wat moet ik mijn zorgverlener vertellen voordat ik EPIVIR inneem?
Vertel uw zorgverlener voordat u EPIVIR 150 mg inneemt als u:
- leverproblemen heeft of heeft gehad, waaronder een infectie met het hepatitis B- of C-virus.
- nierproblemen hebben.
- suikerziekte hebben. Elke dosis van 15 ml (150 mg) EPIVIR drank bevat 3 gram sucrose.
- zwanger bent of van plan bent zwanger te worden. Het gebruik van EPIVIR tijdens de zwangerschap is niet in verband gebracht met een verhoogd risico op geboorteafwijkingen. Praat met uw zorgverlener als u zwanger bent of van plan bent zwanger te worden. Zwangerschapsregister. Er is een zwangerschapsregistratie voor vrouwen die antiretrovirale geneesmiddelen gebruiken tijdens de zwangerschap. Het doel van dit register is om informatie te verzamelen over de gezondheid van u en uw baby. Overleg met uw zorgverlener hoe u kunt deelnemen aan dit register.
- borstvoeding geeft of van plan bent borstvoeding te geven. Geef geen borstvoeding als u EPIVIR gebruikt.
- U mag geen borstvoeding geven als u hiv-1 heeft vanwege het risico dat u hiv-1 op uw baby overdraagt.
Vertel uw zorgverlener over alle medicijnen die u gebruikt, inclusief recept- en vrij verkrijgbare medicijnen, vitamines en kruidensupplementen.
Sommige medicijnen werken samen met EPIVIR. Houd een lijst bij van uw geneesmiddelen en toon deze aan uw zorgverlener en apotheker als u een nieuw geneesmiddel krijgt. U kunt uw zorgverlener of apotheker om een lijst vragen van geneesmiddelen die een wisselwerking hebben met EPIVIR.
Begin niet met het innemen van een nieuw geneesmiddel zonder uw zorgverlener hiervan op de hoogte te stellen. Uw zorgverlener kan u vertellen of het veilig is om EPIVIR samen met andere geneesmiddelen in te nemen.
Hoe moet ik EPIVIR gebruiken?
- Neem EPIVIR precies in zoals uw zorgverlener u zegt dat u het moet innemen.
- Als u een dosis EPIVIR 150 mg bent vergeten, neem deze dan in zodra u eraan denkt. Neem geen 2 doses tegelijk of neem niet meer dan wat uw arts u zegt te nemen.
- Blijf tijdens de behandeling met EPIVIR onder de hoede van een zorgverlener.
- EPIVIR kan met of zonder voedsel worden ingenomen.
- Voor kinderen van 3 maanden en ouder zal uw zorgverlener een dosis EPIVIR 150 mg voorschrijven op basis van het lichaamsgewicht van uw kind.
- Vertel het uw zorgverlener als u of uw kind moeite heeft met het doorslikken van tabletten. EPIVIR 150 mg wordt ook geleverd als een vloeistof (orale oplossing).
- Laat EPIVIR niet opraken. Het virus in uw bloed kan toenemen en het virus kan moeilijker te behandelen worden. Wanneer uw voorraad begint op te raken, haalt u meer bij uw zorgverlener of apotheek.
- Als u te veel EPIVIR 150 mg heeft ingenomen, neem dan onmiddellijk contact op met uw zorgverlener of ga naar de eerste hulpafdeling van het dichtstbijzijnde ziekenhuis.
Wat zijn de mogelijke bijwerkingen van EPIVIR 150 mg?
- EPIVIR 150 mg kan ernstige bijwerkingen veroorzaken, waaronder:
- Zie "Wat is de belangrijkste informatie die ik moet weten over EPIVIR 150 mg?"
- Ophoping van een zuur in uw bloed (melkzuuracidose). Lactaatacidose kan optreden bij sommige mensen die EPIVIR gebruiken. Lactaatacidose is een ernstig medisch noodgeval dat de dood kan veroorzaken. Bel onmiddellijk uw zorgverlener als u een van de volgende symptomen krijgt die kunnen wijzen op lactaatacidose:
- zich erg zwak of moe voelen
- het koud hebben, vooral in je armen en benen
- ongebruikelijke (niet normale) spierpijn
- duizelig of licht in het hoofd voelen
- moeite met ademhalen
- een snelle of onregelmatige hartslag hebben
- maagpijn met misselijkheid en braken
- Ernstige leverproblemen kan optreden bij mensen die EPIVIR gebruiken. In sommige gevallen kunnen deze ernstige leverproblemen tot de dood leiden. Uw lever kan groot worden (hepatomegalie) en u kunt vet in uw lever ontwikkelen (steatose). Bel onmiddellijk uw zorgverlener als u een van de volgende tekenen of symptomen van leverproblemen krijgt:
- uw huid of het witte deel van uw ogen wordt geel (geelzucht)
- verlies van eetlust gedurende meerdere dagen of langer
- misselijkheid
- donkere of "theekleurige" urine
- pijn, pijn of gevoeligheid aan de rechterkant van uw maagstreek
- lichtgekleurde ontlasting (stoelgang)
U heeft een grotere kans op het krijgen van lactaatacidose of ernstige leverproblemen als u een vrouw bent of zeer zwaarlijvig (zwaarlijvig).
- Risico op ontsteking van de alvleesklier (pancreatitis). Kinderen lopen mogelijk risico op het ontwikkelen van pancreatitis tijdens de behandeling met EPIVIR als ze:
- nucleoside-analoge geneesmiddelen heeft gebruikt
- een voorgeschiedenis heeft van pancreatitis in het verleden
- andere risicofactoren hebben voor pancreatitis
Bel onmiddellijk uw zorgverlener als uw kind tekenen en symptomen van pancreatitis ontwikkelt, waaronder ernstige pijn in de bovenbuik, met of zonder misselijkheid en braken. Uw zorgverlener kan u vertellen om te stoppen met het geven van EPIVIR 150 mg aan uw kind als de symptomen en bloedtestresultaten aantonen dat uw kind mogelijk pancreatitis heeft.
- Veranderingen in uw immuunsysteem (immuunreconstitutiesyndroom) kan gebeuren als u hiv-1-geneesmiddelen gaat gebruiken. Uw immuunsysteem kan sterker worden en infecties gaan bestrijden die al lang in uw lichaam verborgen zijn. Vertel het uw zorgverlener meteen als u nieuwe symptomen krijgt nadat u met EPIVIR bent begonnen.
De meest voorkomende bijwerkingen van EPIVIR 150 mg bij volwassenen zijn:
- hoofdpijn
- nasale tekenen en symptomen
- misselijkheid
- diarree
- over het algemeen niet goed voelen
- vermoeidheid
- hoesten
De meest voorkomende bijwerkingen van EPIVIR 150 mg bij kinderen zijn koorts en hoesten.
Vertel het uw zorgverlener als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van EPIVIR. Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
Hoe moet ik EPIVIR bewaren?
- Bewaar EPIVIR 150 mg tabletten en drank bij kamertemperatuur tussen 20 °C en 25 °C (68 °F tot 77 °F).
- Houd flessen EPIVIR 150 mg drank goed gesloten.
Houd EPIVIR en alle geneesmiddelen buiten het bereik van kinderen.
Algemene informatie over het veilige en effectieve gebruik van EPIVIR.
Geneesmiddelen worden soms voorgeschreven voor andere doeleinden dan vermeld in een patiëntenbijsluiter. Gebruik EPIVIR niet voor een aandoening waarvoor het niet is voorgeschreven. Geef EPIVIR niet aan andere mensen, ook niet als zij dezelfde symptomen hebben als u. Het kan hen schaden.
U kunt uw zorgverlener of apotheker om informatie vragen over EPIVIR 150 mg die is geschreven voor gezondheidswerkers.
Ga voor meer informatie naar www.viivhealthcare.com of bel 1-877-844-8872.
Wat zijn de ingrediënten in EPIVIR 150 mg?
Actief bestanddeel: lamivudine
Inactieve ingredienten:
EPIVIR 150 mg filmomhulde tabletten met breukstreep van 150 mg: hypromellose, magnesiumstearaat, microkristallijne cellulose, polyethyleenglycol, polysorbaat 80, natriumzetmeelglycolaat en titaniumdioxide.
EPIVIR 300 mg filmomhulde tabletten: zwart ijzeroxide, hypromellose, magnesiumstearaat, microkristallijne cellulose, polyethyleenglycol, polysorbaat 80, natriumzetmeelglycolaat en titaniumdioxide.
EPIVIR 150 mg drank: kunstmatige aardbeien- en bananensmaken, citroenzuur (watervrij), methylparaben, propyleenglycol, propylparaben, natriumcitraat (dihydraat) en sucrose (200 mg per ml).
Deze patiëntinformatie is goedgekeurd door de Amerikaanse Food and Drug Administration.