Uniphyl 400mg Theophylline Gebruik, bijwerkingen en dosering. Prijs in online apotheek. Generieke medicijnen zonder recept.
Wat is Uniphyl 400 mg en hoe wordt het gebruikt?
Uniphyl is een receptgeneesmiddel dat wordt gebruikt voor de behandeling van de symptomen van astma, bronchitis, emfyseem en andere ademhalingsproblemen (acute bronchospasme). Uniphyl 400 mg kan alleen of in combinatie met andere medicijnen worden gebruikt.
Uniphyl behoort tot een klasse geneesmiddelen die xanthinederivaten worden genoemd; Fosfodiesterase-enzymremmers, niet-selectief.
Het is niet bekend of Uniphyl 400 mg veilig en effectief is bij kinderen jonger dan 1 ½ maand.
Wat zijn de mogelijke bijwerkingen van Uniphyl?
Uniphyl kan ernstige bijwerkingen veroorzaken, waaronder:
- netelroos,
- moeite met ademhalen,
- zwelling van uw gezicht, lippen, tong of keel,
- ernstig of aanhoudend braken,
- aanhoudende hoofdpijn,
- Moeite met slapen,
- snelle hartslagen,
- hartinfarct,
- koorts,
- beenkrampen,
- constipatie,
- onregelmatige hartslagen,
- fladderend in je borst,
- verhoogde dorst of plassen,
- gevoelloosheid of tintelingen,
- spier zwakte,
- slap gevoel,
- verhoogde dorst,
- meer plassen,
- droge mond, en
- fruitige ademgeur
Roep meteen medische hulp in als u een van de bovenstaande symptomen heeft.
De meest voorkomende bijwerkingen van Uniphyl zijn:
- misselijkheid,
- braken,
- diarree,
- hoofdpijn,
- slaapproblemen (slapeloosheid),
- trillingen,
- zweten,
- rusteloosheid, en
- prikkelbaarheid
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van Uniphyl. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
OMSCHRIJVING
Uniphyl (watervrije theofylline-tablet) ® (theofylline, watervrij) Tabletten in een systeem met gecontroleerde afgifte zorgen voor een doseringsinterval van 24 uur voor geschikte patiënten.
Theofylline is structureel geclassificeerd als een methylxanthine. Het komt voor als een wit, geurloos, kristallijn poeder met een bittere smaak.
Watervrij theofylline heeft de chemische naam 1H-Purine-2,6-dion, 3,7-dihydro-1,3-dimethyl-, en wordt weergegeven door de volgende structuurformule:
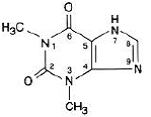
De molecuulformule van watervrije theofylline is C7H8N4O2 met een molecuulgewicht van 180,17.
Elke tablet met gereguleerde afgifte voor orale toediening bevat 400 of 600 mg watervrije theofylline.
Inactieve ingrediënten: cetostearylalcohol, hydroxyethylcellulose, magnesiumstearaat, povidon en talk.
INDICATIES
Theofylline is geïndiceerd voor de behandeling van de symptomen en reversibele luchtwegobstructie geassocieerd met chronische astma en andere chronische longziekten, bijv. emfyseem en chronische bronchitis.
DOSERING EN ADMINISTRATIE
Uniphyl (watervrije theofylline-tablet) 400 of 600 mg tabletten kunnen eenmaal per dag 's morgens of' s avonds worden ingenomen. Het wordt aanbevolen om Uniphyl (watervrije theofylline-tablet) bij de maaltijd in te nemen. Patiënten moeten erop worden gewezen dat als ze ervoor kiezen om Uniphyl (watervrije theofylline-tablet) met voedsel in te nemen, dit consequent met voedsel moet worden ingenomen en dat als ze het in nuchtere toestand innemen, het routinematig op de nuchtere maag moet worden ingenomen. Het is belangrijk dat het product, wanneer het wordt gedoseerd, consistent met of zonder voedsel wordt gedoseerd.
Uniphyl (watervrije theofylline-tablet) ®-tabletten mogen niet worden gekauwd of fijngemaakt omdat dit kan leiden tot een snelle afgifte van theofylline met mogelijk toxiciteit. De tablet met breukstreep kan worden gesplitst. Soms kunnen patiënten die Uniphyl (watervrije theofylline-tablet) 400 of 600 mg tabletten krijgen, een intacte matrixtablet in de ontlasting of via colostoma passeren. Deze matrixtabletten bevatten meestal weinig of geen resterende theofylline.
Gestabiliseerde patiënten van 12 jaar of ouder die een theofyllineproduct met directe afgifte of gereguleerde afgifte gebruiken, kunnen worden overgezet op eenmaal daagse toediening van 400 mg of 600 mg Uniphyl (watervrije theofylline-tablet) mg-basis.
Er moet worden erkend dat de piek- en dalspiegels van theofylline in serum geproduceerd door de eenmaal daagse dosering kunnen verschillen van die geproduceerd door het vorige product en/of regime.
Algemene Overwegingen
De steady-state piekserumtheofyllineconcentratie is een functie van de dosis, het doseringsinterval en de snelheid van theofyllineabsorptie en -klaring bij de individuele patiënt. Vanwege duidelijke individuele verschillen in de snelheid van de theofyllineklaring, varieert de dosis die nodig is om een piekserumconcentratie van theofylline in het bereik van 10-20 mcg/ml te bereiken, viervoudig bij overigens vergelijkbare patiënten bij afwezigheid van factoren waarvan bekend is dat ze de theofyllineklaring veranderen (bijv. 400-1600 mg/dag bij volwassenen De dosis theofylline moet individueel worden bepaald op basis van metingen van de piekserumconcentratie van theofylline om een dosis te bereiken die een maximaal potentieel voordeel oplevert met een minimaal risico op bijwerkingen.
Voorbijgaande cafeïne-achtige bijwerkingen en overmatige serumconcentraties in langzame metaboliseerders kunnen bij de meeste patiënten worden vermeden door te beginnen met een voldoende lage dosis en de dosis langzaam te verhogen, indien klinisch geïndiceerd, in kleine stappen (zie ). Dosisverhogingen mogen alleen worden doorgevoerd als de vorige dosering goed wordt verdragen en met tussenpozen van niet minder dan 3 dagen, zodat de serumtheofyllineconcentraties de nieuwe steady-state bereiken. Dosisaanpassing moet worden geleid door meting van de serumtheofyllineconcentratie (zie: PREVENTIEVE MAATREGELEN , Laboratorium testen en DOSERING EN ADMINISTRATIE ). Zorgverleners dienen patiënten en zorgverleners te instrueren om elke dosering die bijwerkingen veroorzaakt te staken, de medicatie te staken totdat deze symptomen verdwenen zijn en vervolgens de therapie te hervatten met een lagere, eerder getolereerde dosering (zie WAARSCHUWINGEN ).
Als de symptomen van de patiënt goed onder controle zijn, zijn er geen duidelijke bijwerkingen en geen factoren die de doseringsvereisten zouden kunnen veranderen (zie WAARSCHUWINGEN en PREVENTIEVE MAATREGELEN ), moeten serumtheofyllineconcentraties met tussenpozen van 6 maanden worden gecontroleerd voor snelgroeiende kinderen en met jaarlijkse tussenpozen voor alle anderen. Bij acuut zieke patiënten moeten de theofylline-serumconcentraties met regelmatige tussenpozen worden gecontroleerd, bijvoorbeeld elke 24 uur.
Theofylline wordt slecht verdeeld in lichaamsvet, daarom moet de mg/kg-dosis worden berekend op basis van het ideale lichaamsgewicht.
Tabel V bevat het doseringsschema voor theofylline dat wordt aanbevolen voor patiënten in verschillende leeftijdsgroepen en klinische omstandigheden.
Tabel VI bevat aanbevelingen voor dosisaanpassing van theofylline op basis van serumtheofyllineconcentraties. Bij het toepassen van deze algemene doseringsaanbevelingen op individuele patiënten moet rekening worden gehouden met de unieke klinische kenmerken van elke patiënt. Over het algemeen moeten deze aanbevelingen dienen als de bovengrens voor dosisaanpassingen om het risico op mogelijk ernstige bijwerkingen die gepaard gaan met onverwachte grote verhogingen van de serumtheofyllineconcentratie te verminderen.
B. Patiënten met risicofactoren voor verminderde klaring, ouderen (> 60 jaar) en degenen bij wie het niet haalbaar is om de serumtheofyllineconcentraties te controleren:
Bij kinderen van 12-15 jaar mag de theofyllinedosis niet hoger zijn dan 16 mg/kg/dag tot een maximum van 400 mg/dag in aanwezigheid van risicofactoren voor verminderde theofyllineklaring (zie WAARSCHUWINGEN ) of als het niet haalbaar is om de serumtheofyllineconcentraties te controleren.
Bij adolescenten ≥ 16 jaar en volwassenen, inclusief ouderen, mag de dosis theofylline niet hoger zijn dan 400 mg/dag in aanwezigheid van risicofactoren voor verminderde theofyllineklaring (zie WAARSCHUWINGEN ) of als het niet haalbaar is om de serumtheofyllineconcentraties te controleren.
*Patiënten met een sneller metabolisme, klinisch geïdentificeerd door een hoger dan gemiddelde dosisbehoefte, moeten vaker een kleinere dosis krijgen (elke 12 uur) om doorbraaksymptomen als gevolg van lage dalconcentraties vóór de volgende dosis te voorkomen.
TABEL VI. Doseringsaanpassing op basis van serumtheofyllineconcentratie.
HOE GELEVERD
Uniphyl® (theofylline, watervrij) tabletten met gecontroleerde afgifte 400 mg worden geleverd in witte, ondoorzichtige plastic, kindveilige flessen met 100 tabletten ( NDC 67781-251-01) of 500 tabletten ( NDC 67781-251-05). Elke ronde, witte tablet van 400 mg draagt het symbool PF aan de breukstreep en U400 aan de andere kant.
Uniphyl® (theofylline, watervrij) tabletten met gecontroleerde afgifte 600 mg worden geleverd in witte, ondoorzichtige plastic, kindveilige flessen met 100 tabletten ( NDC 67781-252-01). Elke rechthoekige, holle, witte tablet van 600 mg is voorzien van het symbool PF op de breukstreep en U 600 op de andere kant.
Bewaren bij 25°C (77°F); excursies toegestaan tussen 15°-30°C (59°-86°F).
Doseer in een strakke, lichtbestendige container.
Purdue Pharmaceutical Products LP, Dist. door: Purdue Pharmaceutical Products LP, Stamford, CT 06901-3431. 17 maart 2004.
BIJWERKINGEN
Bijwerkingen die verband houden met theofylline zijn over het algemeen mild wanneer de maximale serumtheofyllineconcentraties OVERDOSERING ). De voorbijgaande cafeïne-achtige bijwerkingen treden op bij ongeveer 50% van de patiënten wanneer theofyllinetherapie wordt gestart met doses hoger dan de aanbevolen aanvangsdoses (bijv. > 300 mg/dag bij volwassenen en > 12 mg/kg/dag bij kinderen ouder dan > 1 jaar ). Tijdens de start van de theofyllinetherapie kunnen cafeïne-achtige bijwerkingen het gedrag van de patiënt tijdelijk veranderen, vooral bij schoolgaande kinderen, maar deze reactie houdt zelden aan. Het starten van een behandeling met theofylline met een lage dosis, gevolgd door langzame titratie tot een vooraf bepaalde leeftijdsgebonden maximale dosis, zal de frequentie van deze voorbijgaande bijwerkingen aanzienlijk verminderen (zie DOSERING EN ADMINISTRATIE ). Bij een klein percentage van de patiënten (
Andere bijwerkingen die zijn gemeld bij serumtheofyllineconcentraties
TABEL IV. Manifestaties van theofylline-toxiciteit. *
DRUG-INTERACTIES
Theofylline heeft een wisselwerking met een breed scala aan geneesmiddelen. De interactie kan farmacodynamisch zijn, dwz veranderingen in de therapeutische respons op theofylline of een ander geneesmiddel of het optreden van bijwerkingen zonder een verandering in de serumtheofyllineconcentratie. Vaker echter is de interactie farmacokinetisch, dwz de snelheid van theofyllineklaring wordt veranderd door een ander geneesmiddel, wat resulteert in verhoogde of verlaagde serumtheofyllineconcentraties. Theofylline verandert slechts zelden de farmacokinetiek van andere geneesmiddelen. De in tabel II vermelde geneesmiddelen hebben het potentieel om klinisch significante farmacodynamische of farmacokinetische interacties met theofylline te veroorzaken. De informatie in de kolom "Effect" van tabel II gaat ervan uit dat het interactief werkende geneesmiddel wordt toegevoegd aan een steady-state theofylline-regime. Als theofylline wordt gestart bij een patiënt die al een geneesmiddel gebruikt dat de theofyllineklaring remt (bijv. cimetidine, erytromycine), zal de dosis theofylline die nodig is om een therapeutische serumtheofyllineconcentratie te bereiken, lager zijn. Omgekeerd, als theofylline wordt gestart bij een patiënt die al een geneesmiddel gebruikt dat de theofyllineklaring verbetert (bijv. rifampicine), zal de dosis theofylline die nodig is om een therapeutische serumtheofyllineconcentratie te bereiken, groter zijn. Stopzetting van een gelijktijdig toegediend geneesmiddel dat de klaring van theofylline verhoogt, zal resulteren in accumulatie van theofylline tot potentieel toxische niveaus, tenzij de dosis theofylline op gepaste wijze wordt verlaagd. Stopzetting van een gelijktijdig geneesmiddel dat de klaring van theofylline remt, zal resulteren in verlaagde serumconcentraties van theofylline, tenzij de dosis theofylline op gepaste wijze wordt verhoogd. Van de in tabel III vermelde geneesmiddelen is ofwel gedocumenteerd dat ze geen interactie hebben met theofylline of ze veroorzaken geen klinisch significante interactie (dwz
De lijst van geneesmiddelen in tabellen II en III is actueel vanaf 9 februari 1995. Er worden voortdurend nieuwe interacties gerapporteerd voor theofylline, vooral met nieuwe chemische entiteiten. De beroepsbeoefenaar in de gezondheidszorg mag er niet van uitgaan dat een geneesmiddel geen interactie heeft met theofylline als het niet in tabel II staat. Voordat een nieuw beschikbaar geneesmiddel wordt toegevoegd aan een patiënt die theofylline krijgt, moet de bijsluiter van het nieuwe geneesmiddel en/of de medische literatuur worden geraadpleegd om te bepalen of een interactie tussen het nieuwe geneesmiddel en theofylline is gemeld.
TABEL II. Klinisch significante geneesmiddelinteracties met theofylline.*
TABEL III. Geneesmiddelen waarvan is gedocumenteerd dat ze geen interactie hebben met theofylline of geneesmiddelen die geen klinisch significante interactie met theofylline veroorzaken. *
Geneesmiddel-voedselinteracties
De biologische beschikbaarheid van Uniphyl®-tabletten (theofylline, watervrij) is onderzocht bij gelijktijdige toediening van voedsel. In drie onderzoeken met enkelvoudige doses werden proefpersonen die Uniphyl (watervrije theofylline-tablet) 400 mg of 600 mg tabletten kregen met een gestandaardiseerde vetrijke maaltijd vergeleken met nuchtere omstandigheden. Onder gevoede omstandigheden waren de piekplasmaconcentratie en biologische beschikbaarheid verhoogd; een steile toename van de snelheid en mate van absorptie was echter niet duidelijk (zie: Farmacokinetiek, absorptie ). De verhoogde piek en mate van absorptie onder gevoede omstandigheden suggereert dat de dosering idealiter consistent met of zonder voedsel moet worden toegediend.
Het effect van andere geneesmiddelen op metingen van theofyllineserumconcentraties
De meeste serumtheofylline-assays die klinisch worden gebruikt, zijn immunoassays die specifiek zijn voor theofylline. Andere xanthinen zoals cafeïne, dyphylline en pentoxifylline worden niet gedetecteerd door deze testen. Sommige geneesmiddelen (bijv. cefazoline, cefalothine) kunnen echter interfereren met bepaalde HPLC-technieken. Metabolieten van cafeïne en xanthine bij pasgeborenen of patiënten met nierfunctiestoornissen kunnen ertoe leiden dat de waarde van sommige droge reagensmethoden hoger is dan de werkelijke theofyllineconcentratie in serum.
WAARSCHUWINGEN
Gelijktijdige ziekte
Theofylline moet met uiterste voorzichtigheid worden gebruikt bij patiënten met de volgende klinische aandoeningen vanwege het verhoogde risico op verergering van de gelijktijdige aandoening:
Actieve ulcus pepticum Epileptische aanvallen Hartritmestoornissen (exclusief bradyaritmieën)
Omstandigheden die de theofyllineklaring verminderen
Er zijn verschillende gemakkelijk aanwijsbare oorzaken van verminderde theofyllineklaring. Als de totale dagelijkse dosis niet voldoende wordt verlaagd in aanwezigheid van deze risicofactoren, kan ernstige en mogelijk fatale theofylline-toxiciteit optreden. De voordelen en risico's van het gebruik van theofylline en de noodzaak van intensievere controle van de serumtheofyllineconcentraties bij patiënten met de volgende risicofactoren moeten zorgvuldig worden overwogen:
Leeftijd
Pasgeborenen (voldragen en prematuur) Kinderen 60 jaar)
Gelijktijdige ziekten
Acuut longoedeem Congestief hartfalen Cor-pulmonale koorts; ≥ 102° gedurende 24 uur of langer; of lagere temperatuurstijgingen gedurende langere perioden Hypothyreoïdie Leverziekte; cirrose, acute hepatitis Verminderde nierfunctie bij zuigelingen
Stoppen met roken
Geneesmiddelinteracties
Het toevoegen van een geneesmiddel dat het theofyllinemetabolisme remt (bijv. cimetidine, erytromycine, tacrine) of het stoppen van een gelijktijdig toegediend geneesmiddel dat het theofyllinemetabolisme verbetert (bijv. carbamazepine, rifampicine). (Zien VOORZORGSMAATREGELEN: DRUG-INTERACTIES , ).
Wanneer tekenen of symptomen van theofylline-toxiciteit aanwezig zijn
Wanneer een patiënt die theofylline krijgt misselijkheid of braken ontwikkelt, in het bijzonder herhaaldelijk braken, of andere tekenen of symptomen die overeenkomen met theofylline-toxiciteit (zelfs als een andere oorzaak kan worden vermoed), moeten aanvullende doses theofylline worden onthouden en moet onmiddellijk een serumtheofyllineconcentratie worden gemeten. Patiënten moeten worden geïnstrueerd om niet door te gaan met doseringen die bijwerkingen veroorzaken en om volgende doses te staken totdat de symptomen zijn verdwenen, waarna de beroepsbeoefenaar in de gezondheidszorg de patiënt kan instrueren om het geneesmiddel met een lagere dosering te hervatten (zie DOSERING EN ADMINISTRATIE , Doseringsrichtlijnen, Tabel VI ).
Dosering verhoogt
Verhogingen van de dosis theofylline mogen niet worden gedaan als reactie op een acute verergering van symptomen van chronische longziekte, aangezien theofylline in deze omstandigheden weinig extra voordeel biedt voor geïnhaleerde bèta-2-selectieve agonisten en systemisch toegediende corticosteroïden en het risico op bijwerkingen verhoogt. Een maximale steady-state serumtheofyllineconcentratie moet worden gemeten voordat de dosis wordt verhoogd als reactie op aanhoudende chronische symptomen om vast te stellen of een verhoging van de dosis veilig is. Alvorens de dosis theofylline te verhogen op basis van een lage serumconcentratie, moet de beroepsbeoefenaar in de gezondheidszorg overwegen of het bloedmonster op een geschikt moment in verhouding tot de dosis is afgenomen en of de patiënt zich aan het voorgeschreven regime heeft gehouden (zie PREVENTIEVE MAATREGELEN , Laboratorium testen ).
Aangezien de snelheid van theofyllineklaring dosisafhankelijk kan zijn (dwz de steady-state serumconcentraties kunnen onevenredig stijgen ten opzichte van de dosisverhoging), moet een dosisverhoging op basis van een subtherapeutische serumconcentratiemeting conservatief zijn. Over het algemeen zal het beperken van dosisverhogingen tot ongeveer 25% van de vorige totale dagelijkse dosis het risico op onbedoelde excessieve verhogingen van de serumtheofyllineconcentratie verminderen (zie DOSERING EN ADMINISTRATIE , ).
PREVENTIEVE MAATREGELEN
Algemeen
Een zorgvuldige overweging van de verschillende geneesmiddelen die een wisselwerking hebben en fysiologische aandoeningen die de klaring van theofylline kunnen veranderen en die een aanpassing van de dosering vereisen, moet plaatsvinden vóór de start van de theofylline-therapie, voorafgaand aan verhogingen van de theofylline-dosis en tijdens de follow-up (zie WAARSCHUWINGEN ). De dosis theofylline die is geselecteerd voor het starten van de therapie moet laag zijn en, indien getolereerd, langzaam worden verhoogd over een periode van een week of langer, waarbij de uiteindelijke dosis wordt bepaald door de serumtheofyllineconcentraties en de klinische respons van de patiënt te controleren (zie DOSERING EN ADMINISTRATIE , ).
Controle van serumtheofyllineconcentraties
Metingen van de serumtheofyllineconcentratie zijn direct beschikbaar en moeten worden gebruikt om te bepalen of de dosering geschikt is. Specifiek moet de serumtheofyllineconcentratie als volgt worden gemeten:
Om een dosisverhoging te begeleiden, moet het bloedmonster worden afgenomen op het moment van de verwachte piekserumtheofyllineconcentratie; 12 uur na een avonddosis of 9 uur na een ochtenddosis bij steady-state. Bij de meeste patiënten wordt de steady-state bereikt na 3 dagen toediening als er geen doses zijn overgeslagen, geen extra doses zijn toegevoegd en geen van de doses met ongelijke tussenpozen is ingenomen. Een dalconcentratie (dwz aan het einde van het doseringsinterval) geeft geen aanvullende nuttige informatie en kan leiden tot een ongepaste dosisverhoging, aangezien de maximale serumtheofyllineconcentratie twee of meer keer hoger kan zijn dan de dalconcentratie bij een formulering met onmiddellijke afgifte . Als het serummonster meer dan 12 uur na de avonddosis of meer dan 9 uur na een ochtenddosis wordt afgenomen, moeten de resultaten met voorzichtigheid worden geïnterpreteerd, aangezien de concentratie mogelijk niet overeenkomt met de piekconcentratie. Wanneer daarentegen tekenen of symptomen van theofylline-toxiciteit aanwezig zijn, moet zo snel mogelijk een serummonster worden genomen, onmiddellijk worden geanalyseerd en moet het resultaat onverwijld aan de beroepsbeoefenaar in de gezondheidszorg worden gemeld. Bij patiënten bij wie een verminderde serumeiwitbinding wordt vermoed (bijv. cirrose, vrouwen tijdens het derde trimester van de zwangerschap), moet de concentratie van ongebonden theofylline worden gemeten en moet de dosering worden aangepast om een ongebonden concentratie van 6-12 mcg/ml te bereiken. Speekselconcentraties van theofylline kunnen niet betrouwbaar worden gebruikt om de dosering aan te passen zonder speciale technieken.
Effecten op laboratoriumtests
Als gevolg van de farmacologische effecten verhoogt theofylline bij serumconcentraties binnen het bereik van 10-20 mcg/ml de plasmaglucose enigszins (van gemiddeld 88 mg% tot 98 mg%), urinezuur (van gemiddeld 4 mg/dl). tot 6 mg/dL), vrije vetzuren (van een gemiddelde van 451 μEq/L tot 800 Eq/L, totaal cholesterol (van een gemiddelde van 140 vs 160 mg/dL), HDL (van een gemiddelde van 36 tot 50 mg /dL), HDL/LDL-ratio (van gemiddeld 0,5 tot 0,7), en uitscheiding van vrij cortisol via de urine (van gemiddeld 44 tot 63 mcg/24 uur) Theofylline bij serumconcentraties binnen het bereik van 10-20 mcg/ml kan ook tijdelijk de serumconcentraties van trijoodthyronine verlagen (144 vóór, 131 na één week en 142 ng/dl na 4 weken theofylline).Het klinische belang van deze veranderingen moet worden afgewogen tegen het potentiële therapeutische voordeel van theofylline bij individuele patiënten.
Carcinogenese, mutagenese en verminderde vruchtbaarheid
Carcinogeniteitsonderzoeken op lange termijn zijn uitgevoerd bij muizen (orale doses 30-150 mg/kg) en ratten (orale doses 5-75 mg/kg). Resultaten zijn in afwachting.
Theofylline is onderzocht in Ames salmonella, in vivo en in vitro cytogenetica, micronucleus en ovariumtestsystemen van Chinese hamsters en er is niet aangetoond dat het genotoxisch is.
In een 14 weken durende continue fokstudie, verminderde theofylline, toegediend aan parende paren B6C3F1 muizen in orale doses van 120, 270 en 500 mg/kg (ongeveer 1,0-3,0 maal de dosis voor mensen op basis van mg/m²), de vruchtbaarheid, zoals blijkt uit afname van het aantal levende jongen per nest, afname van het gemiddelde aantal nesten per vruchtbaar paar, en toename van de draagtijd bij de hoge dosis, evenals afname van het aandeel levend geboren jongen bij de middelhoge en hoge dosis. In toxiciteitsonderzoeken van 13 weken werd theofylline toegediend aan F344-ratten en B6C3F1-muizen in orale doses van 40-300 mg/kg (ongeveer 2,0 maal de dosis voor de mens op basis van mg/m²). Bij de hoge dosis werd bij beide soorten systemische toxiciteit waargenomen, waaronder een afname van het gewicht van de testikels.
Zwangerschap
Teratogene effecten: categorie C
In onderzoeken waarbij zwangere muizen, ratten en konijnen werden gedoseerd tijdens de periode van organogenese, veroorzaakte theofylline teratogene effecten.
In onderzoeken met muizen veroorzaakte een enkele intraperitoneale dosis van 100 mg/kg en meer (ongeveer gelijk aan de maximaal aanbevolen orale dosis voor volwassenen op basis van mg/m²) tijdens de organogenese gespleten gehemelte en digitale afwijkingen. Micromelie, micrognathie, klompvoet, subcutaan hematoom, open oogleden en embryoletaliteit werden waargenomen bij doses die ongeveer 2 maal de maximaal aanbevolen orale dosis voor volwassenen waren op basis van mg/m².
In een onderzoek met ratten die werden gedoseerd vanaf de conceptie tot aan de organogenese, veroorzaakte een orale dosis van 150 mg/kg/dag (ongeveer 2 maal de maximaal aanbevolen orale dosis voor volwassenen op basis van mg/m²) digitale afwijkingen. Embryolethaliteit werd waargenomen bij een subcutane dosis van 200 mg/kg/dag (ongeveer 4 keer de maximaal aanbevolen orale dosis voor volwassenen op basis van mg/m²). In een onderzoek waarbij drachtige konijnen gedurende de gehele organogenese werden gedoseerd, werd een intraveneuze dosis van 60 mg/kg/dag (ongeveer 2 maal de maximaal aanbevolen orale dosis voor volwassenen op basis van mg/m²), waarbij één hinde en klinische andere tekenen, produceerde een gespleten gehemelte en was embryoletaal. Doses van en boven 15 mg/kg/dag (minder dan de maximaal aanbevolen orale dosis voor volwassenen op basis van mg/m²) verhoogden de incidentie van skeletvariaties.
Er zijn geen adequate en goed gecontroleerde onderzoeken bij zwangere vrouwen. Theofylline mag alleen tijdens de zwangerschap worden gebruikt als het mogelijke voordeel opweegt tegen het mogelijke risico voor de foetus.
Moeders die borstvoeding geven
Theofylline wordt uitgescheiden in de moedermelk en kan prikkelbaarheid of andere tekenen van milde toxiciteit veroorzaken bij zuigelingen die borstvoeding geven. De concentratie van theofylline in moedermelk is ongeveer gelijk aan de maternale serumconcentratie. Een zuigeling die een liter moedermelk binnenkrijgt die 10-20 mcg/ml theofylline per dag bevat, zal waarschijnlijk 10-20 mg theofylline per dag krijgen. Ernstige bijwerkingen bij de zuigeling zijn onwaarschijnlijk, tenzij de moeder toxische serumtheofyllineconcentraties heeft.
Pediatrisch gebruik
Theofylline is veilig en effectief voor de goedgekeurde indicaties bij pediatrische patiënten. De onderhoudsdosering van theofylline moet met voorzichtigheid worden gekozen bij pediatrische patiënten, aangezien de snelheid van theofyllineklaring zeer variabel is over de pediatrische leeftijdsgroep (zie KLINISCHE FARMACOLOGIE , , , WAARSCHUWINGEN , en DOSERING EN ADMINISTRATIE , ).
Geriatrisch gebruik
Oudere patiënten lopen een significant groter risico op ernstige toxiciteit door theofylline dan jongere patiënten vanwege farmacokinetische en farmacodynamische veranderingen die gepaard gaan met veroudering. De klaring van theofylline is bij gezonde oudere volwassenen (> 60 jaar) met gemiddeld 30% afgenomen in vergelijking met gezonde jonge volwassenen. De klaring van theofylline kan verder worden verminderd door gelijktijdig voorkomende ziekten die voorkomen bij ouderen, die de klaring van dit geneesmiddel verder verslechteren en de serumspiegels en mogelijke toxiciteit kunnen verhogen. Deze aandoeningen omvatten een verminderde nierfunctie, chronische obstructieve longziekte, congestief hartfalen, leverziekte en een verhoogde prevalentie van het gebruik van bepaalde medicijnen (zie PREVENTIEVE MAATREGELEN : DRUG-INTERACTIES ) met het potentieel voor farmacokinetische en farmacodynamische interactie. Eiwitbinding kan verminderd zijn bij ouderen, wat resulteert in een verhoogd aandeel van de totale serumtheofyllineconcentratie in de farmacologisch actieve ongebonden vorm. Oudere patiënten blijken ook gevoeliger te zijn voor de toxische effecten van theofylline na chronische overdosering dan jongere patiënten. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij oudere patiënten (zie: PREVENTIEVE MAATREGELEN , Controle van serumtheofyllineconcentraties , en DOSERING EN ADMINISTRATIE ).
De maximale dagelijkse dosis theofylline bij patiënten ouder dan 60 jaar mag gewoonlijk niet hoger zijn dan 400 mg/dag, tenzij de patiënt symptomatisch blijft en de maximale steady-state serumtheofyllineconcentratie DOSERING EN ADMINISTRATIE ). Bij oudere patiënten dienen theofyllinedoses hoger dan 400 mg/d met voorzichtigheid te worden voorgeschreven.
OVERDOSERING
Algemeen
De chroniciteit en het patroon van overdosering met theofylline hebben een significante invloed op de klinische manifestaties van toxiciteit, het beheer en de uitkomst. Er zijn twee veelvoorkomende presentaties: (1) acute overdosering, dwz inname van een enkele grote overmatige dosis (> 10 mg/kg), zoals optreedt in de context van een zelfmoordpoging of een geïsoleerde medicatiefout, en (2) chronische overdosering, dat wil zeggen, inname van herhaalde doses die buitensporig zijn voor de snelheid van theofyllineklaring door de patiënt. De meest voorkomende oorzaken van chronische overdosering met theofylline zijn onder meer een fout van de patiënt of zorgverlener bij het doseren, een beroepsbeoefenaar in de gezondheidszorg die een te hoge dosis of een normale dosis voorschrijft in aanwezigheid van factoren waarvan bekend is dat ze de snelheid van de theofyllineklaring verminderen, en het verhogen van de dosis als reactie op een exacerbatie van symptomen zonder eerst de serumtheofyllineconcentratie te meten om te bepalen of een dosisverhoging veilig is.
Ernstige toxiciteit door een overdosis theofylline is een relatief zeldzame gebeurtenis. In één zorginstelling was de frequentie van ziekenhuisopnames voor chronische overdosering met theofylline ongeveer 1 per 1000 persoonsjaren blootstelling. In een ander onderzoek, van 6000 bloedmonsters die om welke reden dan ook werden verkregen voor het meten van de serumtheofyllineconcentratie van patiënten die op een afdeling spoedeisende hulp werden behandeld, lag 7% in het bereik van 20-30 mcg/ml en was 3% > 30 mcg/ml. Ongeveer tweederde van de patiënten met serumtheofyllineconcentraties in het bereik van 20-30 mcg/ml had een of meer manifestaties van toxiciteit, terwijl > 90% van de patiënten met serumtheofyllineconcentraties > 30 mcg/ml klinisch bedwelmd waren. Evenzo wordt in andere rapporten ernstige toxiciteit van theofylline voornamelijk gezien bij serumconcentraties > 30 mcg/ml.
Verschillende onderzoeken hebben de klinische manifestaties van een overdosis theofylline beschreven en geprobeerd de factoren te bepalen die levensbedreigende toxiciteit voorspellen. Over het algemeen hebben patiënten die een acute overdosis ervaren minder kans op epileptische aanvallen dan patiënten die een chronische overdosering hebben gehad, tenzij de maximale serumtheofyllineconcentratie > 100 mcg/ml is. Na een chronische overdosering kunnen gegeneraliseerde aanvallen, levensbedreigende hartritmestoornissen en overlijden optreden bij serumtheofyllineconcentraties > 30 mcg/ml. De ernst van de toxiciteit na chronische overdosering is sterker gecorreleerd met de leeftijd van de patiënt dan de piekserumtheofyllineconcentratie; patiënten > 60 jaar lopen het grootste risico op ernstige toxiciteit en mortaliteit na een chronische overdosering. Reeds bestaande of gelijktijdige ziekte kan ook de gevoeligheid van een patiënt voor een bepaalde toxische manifestatie aanzienlijk verhogen, bijv. patiënten met neurologische aandoeningen hebben een verhoogd risico op aanvallen en patiënten met hartaandoeningen hebben een verhoogd risico op hartritmestoornissen voor een gegeven serumtheofyllineconcentratie vergeleken met voor patiënten zonder de onderliggende ziekte.
De frequentie van verschillende gemelde manifestaties van een overdosis theofylline, afhankelijk van de wijze van overdosering, wordt vermeld in tabel IV. Andere manifestaties van theofylline-toxiciteit omvatten verhogingen van serumcalcium, creatinekinase, myoglobine- en leukocytentelling, verlagingen van serumfosfaat en magnesium, acuut myocardinfarct en urineretentie bij mannen met obstructieve uropathie. Aanvallen die gepaard gaan met serumtheofyllineconcentraties > 30 mcg/ml zijn vaak resistent tegen anticonvulsieve therapie en kunnen leiden tot onomkeerbaar hersenletsel als ze niet snel onder controle worden gebracht. Overlijden door theofylline-toxiciteit is meestal secundair aan hartstilstand en/of hypoxische encefalopathie na langdurige gegeneraliseerde aanvallen of hardnekkige hartritmestoornissen die hemodynamisch compromis veroorzaken.
Beheer van overdosis
Algemene aanbevelingen voor patiënten met symptomen van een overdosis theofylline of serumtheofyllineconcentraties > 30 mcg/ml (Opmerking: de serumtheofyllineconcentraties kunnen blijven stijgen na presentatie van de patiënt voor medische zorg.)
Terwijl u tegelijkertijd een behandeling instelt, neemt u contact op met een regionaal antigifcentrum voor bijgewerkte informatie en advies over het individualiseren van de aanbevelingen die volgen.
Stel ondersteunende zorg in, inclusief het instellen van intraveneuze toegang, onderhoud van de luchtwegen en elektrocardiografische monitoring.
Behandeling van aanvallen
Vanwege de hoge morbiditeit en mortaliteit die gepaard gaat met door theofylline geïnduceerde aanvallen, moet de behandeling snel en agressief zijn. Anticonvulsieve therapie moet worden gestart met een intraveneuze benzodiazepine, bijv. diazepam, in stappen van 0,1-0,2 mg/kg elke 1-3 minuten totdat de aanvallen zijn gestopt. Herhaalde aanvallen moeten worden behandeld met een oplaaddosis fenobarbital (20 mg/kg toegediend via een infuus van 30-60 minuten). Case-reports van een overdosis theofylline bij mensen en dierstudies suggereren dat fenytoïne niet effectief is bij het beëindigen van door theofylline veroorzaakte aanvallen. De doses benzodiazepinen en fenobarbital die nodig zijn om de door theofylline geïnduceerde aanvallen te beëindigen, liggen dicht bij de doses die ernstige ademhalingsdepressie of ademstilstand kunnen veroorzaken; de beroepsbeoefenaar in de gezondheidszorg moet daarom bereid zijn om geassisteerde beademing toe te passen. Oudere patiënten en patiënten met COPD kunnen gevoeliger zijn voor de ademhalingsdepressieve effecten van anticonvulsiva. Door barbituraat geïnduceerde coma of toediening van algemene anesthesie kan nodig zijn om herhaalde aanvallen of status epilepticus te beëindigen. Algemene anesthesie moet met voorzichtigheid worden gebruikt bij patiënten met een overdosis theofylline, omdat gefluoreerde vluchtige anesthetica het myocard kunnen sensibiliseren voor endogene catecholamines die door theofylline worden afgegeven. Enfluraan lijkt minder waarschijnlijk geassocieerd te zijn met dit effect dan halothaan en kan daarom veiliger zijn. Neuromusculair blokkerende middelen alleen mogen niet worden gebruikt om aanvallen te beëindigen, aangezien ze de musculoskeletale manifestaties teniet doen zonder de aanvalsactiviteit in de hersenen te beëindigen.
Anticipeer op behoefte aan anticonvulsiva
Bij patiënten met een overdosis theofylline die een hoog risico lopen op theofylline-geïnduceerde aanvallen, bijv. patiënten met acute overdoses en serumtheofyllineconcentraties > 100 mcg/ml of chronische overdosering bij patiënten > 60 jaar met serumtheofyllineconcentraties > 30 mcg/ml , moet worden geanticipeerd op de noodzaak van anticonvulsieve therapie. Een benzodiazepine zoals diazepam moet in een spuit worden opgezogen en bij het bed van de patiënt worden bewaard en medisch personeel dat gekwalificeerd is om aanvallen te behandelen, moet onmiddellijk beschikbaar zijn. Bij geselecteerde patiënten met een hoog risico op door theofylline geïnduceerde aanvallen, dient de toediening van profylactische anticonvulsieve therapie te worden overwogen. Situaties waarin profylactische anticonvulsieve therapie moet worden overwogen bij patiënten met een hoog risico, zijn onder meer verwachte vertragingen bij het instellen van methoden voor extracorporale verwijdering van theofylline (bijv. overdracht van een patiënt met een hoog risico van de ene zorginstelling naar een andere voor extracorporale verwijdering) en klinische omstandigheden die de inspanningen aanzienlijk belemmeren om de theofyllineklaring te verbeteren (bijv. een pasgeborene bij wie dialyse technisch niet haalbaar is of een patiënt met braken die niet reageert op anti-emetica en die niet in staat is meervoudige doses orale actieve kool te verdragen). In dierstudies is aangetoond dat profylactische toediening van fenobarbital, maar niet van fenytoïne, het begin van theofylline-geïnduceerde gegeneraliseerde aanvallen vertraagt en de dosis theofylline verhoogt die nodig is om aanvallen te veroorzaken (dwz de LD50 aanzienlijk verhoogt). Hoewel er geen gecontroleerde studies bij mensen zijn, kan een oplaaddosis van intraveneus fenobarbital (20 mg/kg toegediend via een infuus van 60 minuten) levensbedreigende aanvallen bij risicopatiënten vertragen of voorkomen, terwijl de inspanningen om de theofyllineklaring te verbeteren worden voortgezet. Fenobarbital kan ademhalingsdepressie veroorzaken, vooral bij oudere patiënten en patiënten met COPD.
Behandeling van hartritmestoornissen
Sinustachycardie en eenvoudige ventriculaire premature slagen zijn geen voorbodes van levensbedreigende aritmieën, ze vereisen geen behandeling bij afwezigheid van hemodynamisch compromis, en ze verdwijnen met dalende serumtheofyllineconcentraties. Andere aritmieën, vooral die geassocieerd met hemodynamische stoornissen, moeten worden behandeld met een antiaritmische therapie die geschikt is voor het type aritmie.
Gastro-intestinale decontaminatie
Orale actieve kool (0,5 g/kg tot 20 g en minimaal 1-2 uur na de eerste dosis herhalen) is uiterst effectief in het blokkeren van de absorptie van theofylline door het maagdarmkanaal, zelfs wanneer het enkele uren na inname wordt toegediend. Als de patiënt moet braken, moet de houtskool worden toegediend via een maagsonde of na toediening van een anti-emeticum. Fenothiazine-anti-emetica zoals prochloorperazine of perfenazine moeten worden vermeden, omdat ze de aanvalsdrempel kunnen verlagen en vaak dystonische reacties veroorzaken. Een enkele dosis sorbitol kan worden gebruikt om de stoelgang te bevorderen om de verwijdering van theofylline gebonden aan houtskool uit het maagdarmkanaal te vergemakkelijken. Sorbitol moet echter met voorzichtigheid worden gedoseerd, aangezien het een krachtig zuiverend middel is dat ernstige vocht- en elektrolytenafwijkingen kan veroorzaken, vooral na meerdere doses. In de handel verkrijgbare vaste combinaties van vloeibare houtskool en sorbitol moeten worden vermeden bij jonge kinderen en na de eerste dosis bij adolescenten en volwassenen, aangezien ze geen individualisering van de dosering van houtskool en sorbitol mogelijk maken. Ipecac-siroop moet worden vermeden bij overdoses met theofylline. Hoewel ipecac braken veroorzaakt, vermindert het de absorptie van theofylline niet tenzij het binnen 5 minuten na inname wordt toegediend en zelfs dan is het minder effectief dan orale actieve kool. Bovendien kan door ipecac geïnduceerd braken enkele uren aanhouden na een enkele dosis en de retentie en de effectiviteit van orale actieve kool aanzienlijk verminderen.
Serum Theophy lline
Concentratiebewaking De serumtheofyllineconcentratie moet onmiddellijk na presentatie worden gemeten, 2-4 uur later, en daarna met voldoende tussenpozen, bijv. elke 4 uur, om behandelbeslissingen te sturen en de effectiviteit van de therapie te beoordelen. Serumtheofyllineconcentraties kunnen blijven stijgen na presentatie van de patiënt voor medische zorg als gevolg van voortdurende absorptie van theofylline uit het maagdarmkanaal. Seriële controle van de serumconcentraties van theofylline in serum moet worden voortgezet totdat duidelijk is dat de concentratie niet langer stijgt en is teruggekeerd naar niet-toxische niveaus.
Algemene bewaking
Procedures Elektrocardiografische monitoring moet worden gestart bij presentatie en worden voortgezet totdat de serumtheofyllinespiegel is teruggekeerd naar een niet-toxisch niveau. Serumelektrolyten en glucose moeten worden gemeten bij presentatie en met geschikte tussenpozen die worden aangegeven door klinische omstandigheden. Vloeistof- en elektrolytenafwijkingen moeten onmiddellijk worden gecorrigeerd. Monitoring en behandeling moeten worden voortgezet totdat de serumconcentratie daalt tot onder 20 mcg/ml.
Verbeter de klaring van theofylline
Meervoudige doses orale actieve kool (bijv. 0,5 mg/kg tot 20 g, elke twee uur) verhogen de klaring van theofylline met minstens een factor twee door adsorptie van theofylline uitgescheiden in maag-darmvloeistoffen. Houtskool moet in het maagdarmkanaal worden vastgehouden en er doorheen gaan om effectief te zijn; braken moet daarom onder controle worden gehouden door het toedienen van geschikte anti-emetica. Als alternatief kan de houtskool continu worden toegediend via een maagsonde in combinatie met geschikte anti-emetica. Een enkele dosis sorbitol kan met de actieve kool worden toegediend om de stoelgang te bevorderen en de klaring van de geadsorbeerde theofylline uit het maagdarmkanaal te vergemakkelijken. Sorbitol alleen verbetert de klaring van theofylline niet en moet met voorzichtigheid worden gedoseerd om overmatige ontlasting te voorkomen, wat kan leiden tot ernstige verstoringen van de vocht- en elektrolytenbalans. In de handel verkrijgbare vaste combinaties van vloeibare houtskool en sorbitol moeten worden vermeden bij jonge kinderen en na de eerste dosis bij adolescenten en volwassenen, aangezien ze geen individualisering van de dosering van houtskool en sorbitol mogelijk maken. Bij patiënten met hardnekkig braken moeten extracorporele methoden voor het verwijderen van theofylline worden ingesteld (zie: OVERDOSERING , Buitenlichamelijke verwijdering ).
Specifieke aanbevelingen
Acute overdosis
Chronische overdosering
Buitenlichamelijke verwijdering
Het verhogen van de snelheid van de theofyllineklaring door middel van extracorporale methoden kan de serumconcentraties snel verlagen, maar de risico's van de procedure moeten worden afgewogen tegen het mogelijke voordeel. Houtskoolhemoperfusie is de meest effectieve methode voor extracorporale verwijdering, waarbij de theofyllineklaring tot het zesvoudige wordt verhoogd, maar er kunnen ernstige complicaties optreden, waaronder hypotensie, hypocalciëmie, bloedplaatjesconsumptie en bloedingsdiathesen. Hemodialyse is ongeveer net zo efficiënt als orale actieve kool met meerdere doses en heeft een lager risico op ernstige complicaties dan hemoperfusie met houtskool. Hemodialyse moet als alternatief worden overwogen wanneer hemoperfusie van houtskool niet haalbaar is en orale kool met meervoudige doses niet effectief is vanwege hardnekkig braken. Serumtheofyllineconcentraties kunnen 5-10 mcg / ml terugveren na stopzetting van houtskoolhemoperfusie of hemodialyse als gevolg van herverdeling van theofylline uit het weefselcompartiment. Peritoneale dialyse is niet effectief voor het verwijderen van theofylline; wisseltransfusies bij pasgeborenen waren minimaal effectief.
CONTRA-INDICATIES
Uniphyl (watervrije theofylline-tablet) ® is gecontra-indiceerd bij patiënten met een voorgeschiedenis van overgevoeligheid voor theofylline of andere bestanddelen van het product.
KLINISCHE FARMACOLOGIE
Werkingsmechanisme
Theofylline heeft twee verschillende werkingen in de luchtwegen van patiënten met een omkeerbare obstructie; relaxatie van gladde spieren (dwz bronchodilatatie) en onderdrukking van de respons van de luchtwegen op stimuli (dwz niet-bronchodilaterende profylactische effecten). Hoewel de werkingsmechanismen van theofylline niet met zekerheid bekend zijn, suggereren dierstudies dat bronchodilatatie wordt gemedieerd door de remming van twee isozymen van fosfodiësterase (PDE III en, in mindere mate, PDE IV), terwijl niet-bronchodilaterende profylactische werkingen waarschijnlijk zijn. gemedieerd door een of meer verschillende moleculaire mechanismen, die geen remming van PDE III of antagonisme van adenosinereceptoren inhouden. Sommige van de bijwerkingen die verband houden met theofylline lijken te worden gemedieerd door remming van PDE III (bijv. hypotensie, tachycardie, hoofdpijn en braken) en adenosinereceptorantagonisme (bijv. veranderingen in de cerebrale bloedstroom).
Theofylline verhoogt de contractiekracht van de middenrifspieren. Deze actie lijkt te wijten te zijn aan een verhoging van de calciumopname via een adenosine-gemedieerd kanaal.
Serumconcentratie-effectrelatie
Bronchodilatatie treedt op over het serumtheofyllineconcentratiebereik van 5-20 mcg/ml. In de meeste onderzoeken is een klinisch belangrijke verbetering van de symptoomcontrole gevonden waarvoor maximale serumtheofyllineconcentraties > 10 mcg/ml nodig zijn, maar patiënten met een milde ziekte kunnen baat hebben bij lagere concentraties. Bij serumtheofyllineconcentraties > 20 mcg/ml nemen zowel de frequentie als de ernst van bijwerkingen toe. Over het algemeen zal het handhaven van piekserumtheofyllineconcentraties tussen 10 en 15 mcg/ml het grootste deel van het potentiële therapeutische voordeel van het geneesmiddel opleveren, terwijl het risico op ernstige bijwerkingen wordt geminimaliseerd.
Farmacokinetiek
Overzicht
Theofylline wordt snel en volledig geabsorbeerd na orale toediening in oplossing of vaste orale doseringsvorm met onmiddellijke afgifte. Theofylline ondergaat geen merkbare pre-systemische eliminatie, wordt vrij verdeeld in vetvrije weefsels en wordt uitgebreid gemetaboliseerd in de lever.
De farmacokinetiek van theofylline varieert sterk tussen vergelijkbare patiënten en kan niet worden voorspeld door leeftijd, geslacht, lichaamsgewicht of andere demografische kenmerken. Bovendien kunnen bepaalde gelijktijdige ziekten en veranderingen in de normale fysiologie (zie tabel I) en gelijktijdige toediening van andere geneesmiddelen (zie tabel II) de farmacokinetische kenmerken van theofylline aanzienlijk veranderen. Variabiliteit in het metabolisme binnen de proefpersoon is ook gemeld in sommige onderzoeken, vooral bij acuut zieke patiënten. Het wordt daarom aanbevolen om de serumtheofyllineconcentraties regelmatig te meten bij acuut zieke patiënten (bijv. met tussenpozen van 24 uur) en periodiek bij patiënten die langdurige therapie krijgen, bijv. met tussenpozen van 6-12 maanden. Frequentere metingen moeten worden gedaan in de aanwezigheid van een aandoening die de theofyllineklaring aanzienlijk kan veranderen (zie: VOORZORGSMAATREGELEN, laboratoriumtests ).
TABEL I. Gemiddelde en bereik van totale lichaamsklaring en halfwaardetijd van theofylline gerelateerd aan leeftijd en veranderde fysiologische toestanden.¶
Opmerking: Naast de hierboven genoemde factoren, wordt de theofyllineklaring verhoogd en de halfwaardetijd verlaagd door koolhydraatarme / eiwitrijke diëten, parenterale voeding en dagelijkse consumptie van op houtskool geroosterd rundvlees. Een dieet met veel koolhydraten en weinig eiwitten kan de klaring verminderen en de halfwaardetijd van theofylline verlengen.
Absorptie
Uniphyl (theofylline watervrije tablet) ® toegediend in gevoede toestand wordt volledig geabsorbeerd na orale toediening.
In een cross-overonderzoek met een enkelvoudige dosis werden twee 400 mg Uniphyl (watervrije theofylline-tabletten) 's morgens of' s avonds toegediend aan 19 normale vrijwilligers onmiddellijk na dezelfde gestandaardiseerde maaltijd (769 calorieën bestaande uit 97 gram koolhydraten, 33 gram eiwit en 27 gram dik). Er was geen bewijs van dosisdumping, noch waren er significante verschillen in farmacokinetische parameters die toe te schrijven zijn aan het tijdstip van toediening van het geneesmiddel. In de ochtendarm waren de farmacokinetische parameters AUC = 241,9 ± 83,0 mcg uur/ml, Cmax = 9,3 ± 2,0 mcg/ml, Tmax = 12,8 ± 4,2 uur. In de avondarm waren de farmacokinetische parameters AUC = 219,7 ± 83,0 mcg uur/ml, Cmax = 9,2 ± 2,0 mcg/ml, Tmax = 12,5 ± 4,2 uur.
Een onderzoek waarin Uniphyl (watervrije theofylline-tablet) 400 mg-tabletten werden toegediend aan 17 gevoede volwassen astmapatiënten, produceerde vergelijkbare theofylline-niveau-tijdcurven bij toediening in de ochtend of avond. De serumspiegels waren over het algemeen hoger in het avondregime, maar er waren geen statistisch significante verschillen tussen de twee regimes.
Een onderzoek met een enkele dosis bij 15 normale nuchtere mannelijke vrijwilligers van wie de inherente gemiddelde eliminatiehalfwaardetijd van theofylline werd geverifieerd door een vloeibaar theofyllineproduct van 6,9 ± 2,5 (SD) uur, kreeg twee of drie 400 mg Uniphyl (theofylline watervrije tablet) ® tabletten toegediend . De relatieve biologische beschikbaarheid van Uniphyl (watervrije theofylline-tablet) gegeven in nuchtere toestand in vergelijking met een product met onmiddellijke afgifte was 59%. Piekserumtheofyllinespiegels traden op na 6,9 ± 5,2 (SD) uur, met een genormaliseerd (tot 800 mg) piekniveau van 6,2 ± 2,1 (SD). De schijnbare eliminatiehalfwaardetijd voor de 400 mg Uniphyl (watervrije theofylline-tabletten) tabletten was 17,2 ± 5,8 (SD) uur.
Steady-state farmacokinetiek werd bepaald in een onderzoek bij 12 nuchtere patiënten met chronische reversibele obstructieve longziekte. Alle werden gedoseerd met twee 400 mg Uniphyl (watervrije theofylline-tabletten) eenmaal daags 's ochtends en een BID-referentieproduct met gecontroleerde afgifte toegediend als twee tabletten van 200 mg met een tussenpoos van 12 uur. De farmacokinetische parameters die werden verkregen voor Uniphyl (watervrije theofylline-tablet) tabletten gegeven in doses van 800 mg eenmaal daags 's morgens waren vrijwel identiek aan de overeenkomstige parameters voor het referentiegeneesmiddel bij toediening als 400 mg tweemaal daags. In het bijzonder waren de AUC-, Cmax- en Cmin-waarden die in dit onderzoek werden verkregen als volgt:
Studies met enkelvoudige doses waarbij proefpersonen twaalf (12) uur vóór en nog eens vier (4) uur na toediening vasten, lieten een verminderde biologische beschikbaarheid zien in vergelijking met dosering met voedsel. Een studie met een enkelvoudige dosis bij 20 normale vrijwilligers die twee (2) tabletten van 400 mg 's ochtends kregen, vergeleek de dosering onder deze nuchtere omstandigheden met de dosering direct voorafgaand aan een gestandaardiseerd ontbijt (769 calorieën, bestaande uit 97 gram koolhydraten, 33 gram eiwit en 27 gram vet). Onder gevoede omstandigheden waren de farmacokinetische parameters: AUC = 231,7 ± 92,4 mcg uur/ml, Cmax = 8,4 ± 2,6 mcg/ml, Tmax = 17,3 ± 6,7 uur. Onder nuchtere omstandigheden waren deze parameters AUC = 141,2 ± 6,53 mcg uur/ml, Cmax = 5,5 ± 1,5 mcg/ml, Tmax = 6,5 ± 2,1 uur.
Een ander onderzoek met een enkelvoudige dosis bij 21 normale mannelijke vrijwilligers, 's avonds gedoseerd, vergeleek vasten met een gestandaardiseerde maaltijd met veel calorieën en veel vet (870-1.020 calorieën, bestaande uit 33 gram eiwit, 55-75 gram vet, 58 gram koolhydraten). In de nuchtere arm kregen de proefpersonen één Uniphyl (theofylline watervrije tablet) ® 400 mg tablet om 20.00 uur na acht uur vasten, gevolgd door nog eens vier uur vasten. In de gevoede arm kregen de proefpersonen opnieuw één 400 mg Uniphyl (watervrije theofylline-tablet) tablet, maar om 20.00 uur onmiddellijk na de hierboven genoemde gestandaardiseerde maaltijd met een hoog vetgehalte. De farmacokinetische parameters (genormaliseerd naar 800 mg) bij eten waren AUC = 221,8 ± 40,9 mcg uur/ml, Cmax = 10,9 ± 1,7 mcg/ml, Tmax = 11,8 ± 2,2 uur. In de nuchtere arm waren de farmacokinetische parameters (genormaliseerd naar 800 mg) AUC = 146,4 ± 40,9 mcg uur/ml, Cmax = 6,7 ± 1,7 mcg/ml, Tmax = 7,3 ± 2,2 uur.
Toediening van enkelvoudige doses Uniphyl (watervrije theofylline-tablet) aan gezonde, normale vrijwilligers, onder langdurige vastenomstandigheden (ten minste 10 uur 's nachts vasten voor dosering gevolgd door nog eens vier (4) uur vasten na dosering) resulteert in verminderde biologische beschikbaarheid. Er was echter geen storing in dit toedieningssysteem dat leidde tot een plotselinge en onverwachte afgifte van een grote hoeveelheid theofylline met Uniphyl (watervrije theofylline-tablet) tabletten, zelfs wanneer ze worden toegediend met een vetrijke, calorierijke maaltijd.
Vergelijkbare onderzoeken werden uitgevoerd met de 600 mg Uniphyl (watervrije theofylline-tablet) tablet. Een onderzoek met een enkelvoudige dosis bij 24 proefpersonen met een vastgestelde theofyllineklaring van ≤ 4 l/uur, vergeleek de farmacokinetische evaluatie van één 600 mg Uniphyl (watervrije theofylline tablet) tablet en anderhalve 400 mg Uniphyl (theofylline watervrije tablet) tabletten onder gevoed (met behulp van een standaard vetrijk dieet) en nuchtere omstandigheden. De resultaten van deze 4-weg gerandomiseerde cross-over studie tonen de bio-equivalentie aan van de 400 mg en 600 mg Uniphyl (theofylline watervrije tablet) tabletten. Onder gevoede omstandigheden waren de farmacokinetische resultaten voor de anderhalve 400 mg tabletten AUC = 214,64 ± 55,88 mcg uur/ml, Cmax = 10,58 ± 2,21 mcg/ml en Tmax = 9,00 ± 2,64 uur, en voor de 600 mg tablet waren AUC = 207,85 ± 48,9 mcg uur/ml, Cmax = 10,39 ± 1,91 mcg/ml en Tmax = 9,58 ± 1,86 uur. In nuchtere toestand waren de farmacokinetische resultaten voor de anderhalve 400 mg tabletten AUC = 191,85 ± 51,1 mcg uur/ml, Cmax = 7,37 ± 1,83 mcg/ml en Tmax = 8,08 ± 4,39 uur; en voor de 600 mg tablet waren AUC = 199,39 ± 70,27 mcg uur/ml, Cmax = 7,66 ± 2,09 mcg/ml en Tmax = 9,67 ± 4,89 uur.
In deze studie waren de gemiddelde verhoudingen van gevoed/nuchter voor de anderhalve tablet van 400 mg en de tablet van 600 mg respectievelijk ongeveer 112% en 104%.
In een ander onderzoek werd de biologische beschikbaarheid van de 600 mg Uniphyl (watervrije theofylline-tablet) tablet onderzocht bij toediening 's ochtends en 's avonds. Dit cross-overonderzoek met een enkelvoudige dosis bij 22 gezonde mannen werd uitgevoerd onder gevoede omstandigheden (standaard vetrijk dieet). De resultaten toonden geen klinisch significant verschil aan in de biologische beschikbaarheid van de 600 mg Uniphyl (watervrije theofylline-tablet) die 's ochtends of' s avonds werd toegediend. De resultaten waren: AUC = 233,6 ± 45,1 mcg uur/ml, Cmax = 10,6 ± 1,3 mcg/ml en Tmax = 12,5 ± 3,2 uur bij ochtenddosering; AUC = 209,8 ± 46,2 mcg uur/ml, Cmax = 9,7 ± 1,4 mcg/ml en Tmax = 13,7 ± 3,3 uur bij avonddosering. De PM/AM-ratio bedroeg 89,3%.
De absorptie-eigenschappen van Uniphyl®-tabletten (theofylline, watervrij) zijn uitgebreid bestudeerd. Een steady-state cross-over biologische beschikbaarheidsstudie bij 22 normale mannen vergeleek twee Uniphyl (watervrije theofylline-tabletten) 400 mg tabletten die elke 24 uur om 8 uur 's ochtends onmiddellijk na het ontbijt werden toegediend met een referentie-theofylline-product met gecontroleerde afgifte dat tweemaal daags werd toegediend aan gevoede proefpersonen om 8 uur onmiddellijk na het ontbijt en 20.00 uur direct na het avondeten (769 calorieën, bestaande uit 97 gram koolhydraten, 33 gram eiwit en 27 gram vet).
De farmacokinetische parameters voor Uniphyl (theofylline watervrije tablet) 400 mg tabletten onder deze steady-state omstandigheden waren AUC = 203,3 ± 87,1 mcg uur/ml, Cmax = 12,1 ± 3,8 mcg/ml, Cmin = 4,50 ± 3,6, Tmax = 8,8 ± 4,6 uur. Voor het BID-referentieproduct waren de farmacokinetische parameters AUC = 219,2 ± 88,4 mcg uur/ml, Cmax = 11,0 ± 4,1 mcg/ml, Cmin = 7,28 ± 3,5, Tmax = 6,9 ± 3,4 uur. De gemiddelde procentuele fluctuatie [(Cmax-Cmin/Cmin)x100] = 169% voor het eenmaal daagse regime en 51% voor het referentieproduct BID-regime.
De biologische beschikbaarheid van de 600 mg Uniphyl (watervrije theofylline-tablet) tablet werd verder geëvalueerd in een steady-state-onderzoek met meerdere doses bij 26 gezonde mannen waarin de 600 mg-tablet werd vergeleken met anderhalve 400 mg Uniphyl (theofylline watervrije tablet) tabletten. Alle proefpersonen hadden eerder een theofyllineklaring van ≤ 4 l/uur vastgesteld en werden gedurende 6 dagen eenmaal daags toegediend onder gevoede omstandigheden. De resultaten toonden geen klinisch significant verschil tussen de 600 mg en de anderhalve 400 mg Uniphyl (theofylline watervrije tablet) tabletregimes. Steady-state resultaten waren:
De biologische beschikbaarheidsratio voor de 600/400 mg tabletten was 98,8%. Onder alle studieomstandigheden is de tablet van 600 mg dus bio-equivalent aan anderhalve tablet van 400 mg.
Studies tonen aan dat, zolang proefpersonen ofwel consequent werden gevoed of consequent vasten, er een vergelijkbare biologische beschikbaarheid is met eenmaal daagse toediening van Uniphyl (watervrije theofylline-tabletten) tabletten, ongeacht of ze 's morgens of' s avonds worden gedoseerd.
Verdeling
Zodra theofylline in de systemische circulatie komt, wordt ongeveer 40% gebonden aan plasma-eiwit, voornamelijk albumine. Ongebonden theofylline verspreidt zich door het lichaamswater, maar wordt slecht verdeeld in lichaamsvet. Het schijnbare distributievolume van theofylline is ongeveer 0,45 l/kg (bereik 0,3-0,7 l/kg) gebaseerd op het ideale lichaamsgewicht. Theofylline passeert vrijelijk door de placenta, in de moedermelk en in de cerebrospinale vloeistof (CSF). Speekseltheofyllineconcentraties benaderen ongebonden serumconcentraties, maar zijn niet betrouwbaar voor routinematige of therapeutische controle tenzij speciale technieken worden gebruikt. Een toename van het distributievolume van theofylline, voornamelijk als gevolg van een afname van de plasma-eiwitbinding, treedt op bij premature neonaten, patiënten met levercirrose, ongecorrigeerde acidemie, ouderen en bij vrouwen tijdens het derde trimester van de zwangerschap. In dergelijke gevallen kan de patiënt tekenen van toxiciteit vertonen bij totale (gebonden+ongebonden) serumconcentraties van theofylline in het therapeutische bereik (10-20 mcg/ml) als gevolg van verhoogde concentraties van het farmacologisch actieve ongebonden geneesmiddel. Evenzo kan een patiënt met verminderde theofyllinebinding een subtherapeutische totale geneesmiddelconcentratie hebben, terwijl de farmacologisch actieve ongebonden concentratie in het therapeutische bereik ligt. Als alleen de totale serumtheofyllineconcentratie wordt gemeten, kan dit leiden tot een onnodige en mogelijk gevaarlijke dosisverhoging. Bij patiënten met een verminderde eiwitbinding is het meten van de ongebonden serumtheofyllineconcentratie een betrouwbaarder manier om de dosering aan te passen dan het meten van de totale serumtheofyllineconcentratie. Over het algemeen moeten concentraties van ongebonden theofylline binnen het bereik van 6-12 mcg/ml worden gehouden.
Metabolisme
Na orale toediening ondergaat theofylline geen meetbare first-pass eliminatie. Bij volwassenen en kinderen ouder dan één jaar wordt ongeveer 90% van de dosis in de lever gemetaboliseerd. Biotransformatie vindt plaats door demethylering tot 1-methylxanthine en 3-methylxanthine en hydroxylering tot 1,3-dimethylurinezuur. 1-methylxanthine wordt verder gehydroxyleerd door xanthine-oxidase tot 1-methylurinezuur. Ongeveer 6% van een dosis theofylline is N-gemethyleerd tot cafeïne. Demethylering van theofylline tot 3-methylxanthine wordt gekatalyseerd door cytochroom P-450 1A2, terwijl cytochromen P-450 2E1 en P-450 3A3 de hydroxylering tot 1,3-dimethylurinezuur katalyseren. Demethylering tot 1-methylxanthine lijkt te worden gekatalyseerd door cytochroom P-450 1A2 of een nauw verwant cytochroom. Bij pasgeborenen is de N-demethyleringsroute afwezig, terwijl de functie van de hydroxyleringsroute duidelijk gebrekkig is. De activiteit van deze routes neemt langzaam toe tot het maximale niveau op de leeftijd van één jaar.
Cafeïne en 3-methylxanthine zijn de enige theofyllinemetabolieten met farmacologische activiteit. 3-methylxanthine heeft ongeveer een tiende van de farmacologische activiteit van theofylline en serumconcentraties bij volwassenen met een normale nierfunctie zijn
Zowel de N-demethylerings- als hydroxyleringsroutes van theofylline-biotransformatie zijn beperkt in capaciteit. Vanwege de grote interindividuele variabiliteit van de snelheid van het theofyllinemetabolisme, kan bij sommige patiënten non-lineariteit van eliminatie beginnen bij serumtheofyllineconcentraties DOSERING EN TOEDIENING, Tabel VI ). Nauwkeurige voorspelling van de dosisafhankelijkheid van het theofyllinemetabolisme bij patiënten a priori is niet mogelijk, maar patiënten met zeer hoge initiële klaringspercentages (dwz lage steady-state serumtheofyllineconcentraties bij bovengemiddelde doses) hebben de grootste kans op het ervaren van grote veranderingen in serum theofyllineconcentratie als reactie op doseringswijzigingen.
uitscheiding
Bij pasgeborenen wordt ongeveer 50% van de theofyllinedosis onveranderd in de urine uitgescheiden. Na de eerste drie levensmaanden wordt ongeveer 10% van de theofyllinedosis onveranderd in de urine uitgescheiden. De rest wordt voornamelijk in de urine uitgescheiden als 1,3-dimethylurinezuur (35-40%), 1-methylurinezuur (20-25%) en 3-methylxanthine (15-20%). Aangezien weinig theofylline onveranderd in de urine wordt uitgescheiden en aangezien actieve metabolieten van theofylline (dwz cafeïne, 3-methylxanthine) zich niet ophopen tot klinisch significante niveaus, zelfs niet bij een terminale nierziekte, is geen dosisaanpassing voor nierinsufficiëntie nodig bij volwassenen en kinderen > 3 maanden. Daarentegen vereist de grote fractie van de theofyllinedosis die in de urine wordt uitgescheiden als onveranderde theofylline en cafeïne bij pasgeborenen, zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties bij pasgeborenen met een verminderde nierfunctie (zie WAARSCHUWINGEN ).
Serumconcentraties bij Steady-State
Na meerdere doses theofylline wordt bij volwassenen een steady-state bereikt in 30-65 uur (gemiddeld 40 uur). Bij steady-state, bij een doseringsschema met tussenpozen van 24 uur, is de verwachte gemiddelde dalconcentratie ongeveer 50% van de gemiddelde piekconcentratie, uitgaande van een gemiddelde theofyllinehalfwaardetijd van 8 uur. Het verschil tussen piek- en dalconcentraties is groter bij patiënten met een snellere theofyllineklaring. Bij deze patiënten kan toediening van Uniphyl (watervrije theofylline-tablet)® vaker nodig zijn (elke 12 uur).
Speciale populaties (zie tabel I voor gemiddelde klaring en halfwaardetijd)
geriatrische
De klaring van theofylline is bij gezonde oudere volwassenen (> 60 jaar) met gemiddeld 30% afgenomen in vergelijking met gezonde jonge volwassenen. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij oudere patiënten (zie: WAARSCHUWINGEN ).
Kindergeneeskunde
De klaring van theofylline is erg laag bij pasgeborenen (zie: WAARSCHUWINGEN ). De theofyllineklaring bereikt maximale waarden op de leeftijd van één jaar, blijft relatief constant tot ongeveer 9 jaar en neemt dan langzaam af met ongeveer 50% tot volwassen waarden rond de leeftijd van 16 jaar. De renale excretie van onveranderd theofylline bij pasgeborenen bedraagt ongeveer 50% van de dosis, vergeleken met ongeveer 10% bij kinderen ouder dan drie maanden en bij volwassenen. Bij pediatrische patiënten is zorgvuldige aandacht voor de doseringskeuze en monitoring van de serumtheofyllineconcentraties vereist (zie: WAARSCHUWINGEN en DOSERING EN ADMINISTRATIE ).
Geslacht
Geslachtsverschillen in de klaring van theofylline zijn relatief klein en het is onwaarschijnlijk dat ze van klinische betekenis zijn. Een significante vermindering van de theofyllineklaring is echter gemeld bij vrouwen op de 20e dag van de menstruatiecyclus en tijdens het derde trimester van de zwangerschap.
Ras
Farmacokinetische verschillen in theofyllineklaring als gevolg van ras zijn niet onderzocht.
Nierinsufficiëntie
Slechts een kleine fractie, bijvoorbeeld ongeveer 10%, van de toegediende dosis theofylline wordt onveranderd uitgescheiden in de urine van kinderen ouder dan drie maanden en volwassenen. Aangezien weinig theofylline onveranderd in de urine wordt uitgescheiden en aangezien actieve metabolieten van theofylline (dwz cafeïne, 3-methylxanthine) zich niet ophopen tot klinisch significante niveaus, zelfs niet bij een nierziekte in het eindstadium, is er geen dosisaanpassing nodig voor nierinsufficiëntie bij volwassenen en kinderen > 3 maanden. Daarentegen wordt bij pasgeborenen ongeveer 50% van de toegediende dosis theofylline onveranderd in de urine uitgescheiden. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij pasgeborenen met een verminderde nierfunctie (zie WAARSCHUWINGEN ).
Leverinsufficiëntie
De theofyllineklaring is met 50% of meer verminderd bij patiënten met leverinsufficiëntie (bijv. cirrose, acute hepatitis, cholestase). Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij patiënten met een verminderde leverfunctie (zie WAARSCHUWINGEN ).
Congestief hartfalen (CHF)
De theofyllineklaring is met 50% of meer verlaagd bij patiënten met CHF. De mate van vermindering van de theofyllineklaring bij patiënten met CHF lijkt direct gecorreleerd te zijn met de ernst van de hartziekte. Aangezien de theofyllineklaring onafhankelijk is van de bloedstroom in de lever, lijkt de afname van de klaring eerder te wijten te zijn aan een verminderde hepatocytfunctie dan aan een verminderde perfusie. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij patiënten met CHF (zie: WAARSCHUWINGEN ).
Rokers
Het roken van tabak en marihuana lijkt de klaring van theofylline te verhogen door inductie van metabole routes. Het is aangetoond dat de theofyllineklaring met ongeveer 50% toeneemt bij jonge volwassen tabaksrokers en met ongeveer 80% bij oudere tabaksrokers in vergelijking met niet-rokers. Van passieve rookblootstelling is ook aangetoond dat het de theofyllineklaring tot 50% verhoogt. Onthouding van het roken van tabak gedurende één week veroorzaakt een vermindering van ongeveer 40% van de theofyllineklaring. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij patiënten die stoppen met roken (zie: WAARSCHUWINGEN ). Het is aangetoond dat het gebruik van nicotinegom geen effect heeft op de theofyllineklaring.
Koorts
Koorts, ongeacht de onderliggende oorzaak, kan de klaring van theofylline verminderen. De omvang en duur van de koorts lijken direct gecorreleerd te zijn met de mate van afname van de theofyllineklaring. Precieze gegevens ontbreken, maar een temperatuur van 39°C (102°F) gedurende ten minste 24 uur is waarschijnlijk vereist om een klinisch significante verhoging van de serumtheofyllineconcentraties te veroorzaken. Kinderen met een snelle theofyllineklaring (dwz degenen die een dosis nodig hebben die aanzienlijk hoger is dan gemiddeld [bijv. > 22 mg/kg/dag] om een therapeutische piekserumtheofyllineconcentratie te bereiken wanneer ze koorts hebben) kunnen een groter risico lopen op toxische effecten van verminderde klaring tijdens aanhoudende koorts. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij patiënten met aanhoudende koorts (zie: WAARSCHUWINGEN ).
Diversen
Andere factoren die verband houden met verminderde theofyllineklaring zijn het derde trimester van de zwangerschap, sepsis met meervoudig orgaanfalen en hypothyreoïdie. Zorgvuldige aandacht voor dosisverlaging en frequente controle van de serumtheofyllineconcentraties zijn vereist bij patiënten met een van deze aandoeningen (zie WAARSCHUWINGEN ). Andere factoren die in verband worden gebracht met een verhoogde theofyllineklaring zijn onder meer hyperthyreoïdie en cystische fibrose.
Klinische studies
Bij patiënten met chronisch astma, waaronder patiënten met ernstig astma die inhalatiecorticosteroïden of om de dag orale corticosteroïden nodig hebben, hebben veel klinische onderzoeken aangetoond dat theofylline de frequentie en ernst van symptomen vermindert, waaronder nachtelijke exacerbaties, en het "indien nodig" gebruik van inhalatie bèta-2-agonisten. Het is ook aangetoond dat theofylline de behoefte aan korte kuren met dagelijkse orale prednison vermindert om exacerbaties van luchtwegobstructie die niet reageren op bronchodilatoren bij astmapatiënten te verlichten.
Bij patiënten met chronische obstructieve longziekte (COPD) hebben klinische onderzoeken aangetoond dat theofylline dyspnoe, luchtinsluiting en het ademen vermindert en de contractiliteit van de middenrifspieren verbetert met weinig of geen verbetering van de longfunctiemetingen.
PATIËNT INFORMATIE
De patiënt (of ouder/verzorger) moet worden geïnstrueerd om medisch advies in te winnen wanneer misselijkheid, braken, aanhoudende hoofdpijn, slapeloosheid of snelle hartslag optreden tijdens de behandeling met theofylline, zelfs als een andere oorzaak wordt vermoed. De patiënt moet worden geïnstrueerd om contact op te nemen met zijn/haar beroepsbeoefenaar in de gezondheidszorg als hij een nieuwe ziekte ontwikkelt, vooral als deze gepaard gaat met aanhoudende koorts, als hij een verergering van een chronische ziekte ervaart, als hij begint of stopt met het roken van sigaretten of marihuana, of als een andere beroepsbeoefenaar in de gezondheidszorg een nieuw medicijn of het stopzetten van een eerder voorgeschreven medicijn. Patiënten moeten worden geïnformeerd dat theofylline een wisselwerking heeft met een grote verscheidenheid aan geneesmiddelen (zie tabel II). Het voedingssupplement sint-janskruid (Hypericum perforatum) mag niet tegelijkertijd met theofylline worden ingenomen, omdat dit kan leiden tot verlaagde theofyllinespiegels. Als patiënten al sint-janskruid en theofylline samen gebruiken, moeten ze hun arts raadplegen voordat ze stoppen met het gebruik van sint-janskruid, aangezien hun theofyllineconcentraties kunnen stijgen wanneer dit wordt gedaan, wat kan leiden tot toxiciteit. Patiënten moeten de instructie krijgen om alle beroepsbeoefenaren in de gezondheidszorg die bij hun zorg betrokken zijn, te informeren dat ze theofylline gebruiken, vooral wanneer een medicijn wordt toegevoegd aan of verwijderd uit hun behandeling. Patiënten moeten worden geïnstrueerd om de dosis, het tijdstip van de dosis of de toedieningsfrequentie niet te wijzigen zonder eerst hun zorgverlener te raadplegen. Als een dosis wordt gemist, moet de patiënt worden geïnstrueerd om de volgende dosis op het gewoonlijk geplande tijdstip in te nemen en niet te proberen de gemiste dosis in te halen.
Uniphyl (theofylline watervrije tablet) ® Tabletten kunnen eenmaal per dag 's ochtends of' s avonds worden ingenomen. Het wordt aanbevolen om Uniphyl (watervrije theofylline-tablet) bij de maaltijd in te nemen. Patiënten moeten erop worden gewezen dat als ze ervoor kiezen om Uniphyl (watervrije theofylline-tablet) met voedsel in te nemen, dit consequent met voedsel moet worden ingenomen en dat als ze het in nuchtere toestand innemen, het routinematig op de nuchtere maag moet worden ingenomen. Het is belangrijk dat het product, wanneer het wordt gedoseerd, consistent met of zonder voedsel wordt gedoseerd.
Uniphyl (watervrije theofylline-tablet) Tabletten mogen niet worden gekauwd of fijngemaakt omdat dit kan leiden tot een snelle afgifte van theofylline met mogelijk toxiciteit. De tablet met breukstreep kan worden gesplitst. Patiënten die Uniphyl (watervrije theofylline-tablet) krijgen, kunnen een intacte matrixtablet in de ontlasting of via colostoma passeren. Deze matrixtabletten bevatten meestal weinig of geen resterende theofylline.