Xeloda 500mg Capecitabine Gebruik, bijwerkingen en dosering. Prijs in online apotheek. Generieke medicijnen zonder recept.
Wat is Xeloda 500 mg en hoe wordt het gebruikt?
Xeloda is een receptgeneesmiddel dat wordt gebruikt voor de behandeling van de symptomen van kankers zoals darmkanker, colorectale kanker en borstkanker. Xeloda kan alleen of in combinatie met andere medicijnen worden gebruikt.
Xeloda behoort tot een klasse geneesmiddelen die antineoplastische middelen, antimetaboliet, worden genoemd.
Het is niet bekend of Xeloda 500 mg veilig en effectief is bij kinderen.
Wat zijn de mogelijke bijwerkingen van Xeloda 500 mg?
Xeloda kan ernstige bijwerkingen veroorzaken, waaronder:
- koorts boven 100,5 graden,
- misselijkheid,
- verlies van eetlust,
- veel minder eten dan normaal,
- braken (meer dan eens per 24 uur),
- ernstige diarree (meer dan 4 keer per dag of 's nachts),
- blaren of zweren in uw mond,
- rood of gezwollen tandvlees,
- moeite met slikken,
- pijn, gevoeligheid, roodheid, zwelling, blaarvorming of vervelling van de huid van uw handen of voeten,
- erg dorstig of warm voelen,
- niet kunnen plassen,
- zwaar zweten,
- hete en droge huid,
- pijn op de borst of druk,
- ongelijke hartslagen,
- kortademigheid,
- zwelling of snelle gewichtstoename,
- pijnlijk of moeilijk plassen,
- zwelling in uw voeten of enkels,
- zich moe voelen,
- kortademigheid,
- donkere urine,
- kleikleurige ontlasting,
- geel worden van de huid of ogen (geelzucht),
- koorts of andere griepsymptomen,
- hoesten,
- huidzweren,
- bleke huid,
- gemakkelijk blauwe plekken,
- ongewone bloeding,
- licht in het hoofd voelen,
- snelle hartslag,
- keelpijn,
- zwelling in uw gezicht of tong,
- branden in je ogen, en
- huidpijn die wordt gevolgd door een rode of paarse uitslag (vooral het gezicht of het bovenlichaam) en blaarvorming en vervelling veroorzaakt
Roep meteen medische hulp in als u een van de bovenstaande symptomen heeft.
De meest voorkomende bijwerkingen van Xeloda 500 mg zijn:
- buikpijn,
- constipatie,
- buikpijn,
- moe gevoel,
- milde huiduitslag, en
- gevoelloosheid of tintelingen in uw handen of voeten
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van Xeloda. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
WAARSCHUWING
XELODA-WARFARIN INTERACTIE
Interactie met XELODA 500 mg warfarine: Bij patiënten die gelijktijdig met capecitabine en orale cumarine-derivaat anticoagulantia worden behandeld, moet hun anticoagulansrespons (INR of protrombinetijd) regelmatig worden gecontroleerd om de dosis anticoagulantia dienovereenkomstig aan te passen. Een klinisch belangrijke geneesmiddelinteractie tussen XELODA en warfarine werd aangetoond in een klinisch farmacologisch onderzoek [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en DRUG-INTERACTIES]. Veranderde stollingsparameters en/of bloedingen, waaronder overlijden, zijn gemeld bij patiënten die XELODA 500 mg gelijktijdig gebruikten met cumarinederivaten zoals warfarine en fenprocoumon. Postmarketingrapporten hebben klinisch significante verhogingen van de protrombinetijd (PT) en INR aangetoond bij patiënten die gestabiliseerd waren op anticoagulantia op het moment dat XELODA 500 mg werd geïntroduceerd. Deze voorvallen traden op binnen enkele dagen en tot enkele maanden na het starten van de behandeling met XELODA 500 mg en, in enkele gevallen, binnen 1 maand na het stoppen met XELODA. Deze voorvallen traden op bij patiënten met en zonder levermetastasen. Leeftijd ouder dan 60 jaar en een diagnose van kanker maken patiënten onafhankelijk vatbaar voor een verhoogd risico op coagulopathie.
OMSCHRIJVING
XELODA (capecitabine) is een fluoropyrimidinecarbamaat met antineoplastische activiteit. Het is een oraal toegediende systemische prodrug van 5'-deoxy-5-fluorouridine (5'-DFUR) die wordt omgezet in 5-fluorouracil.
De chemische naam voor capecitabine is 5'-deoxy-5-fluor-N-[(pentyloxy)carbonyl]-cytidine en heeft een molecuulgewicht van 359,35. Capecitabine heeft de volgende structuurformule:
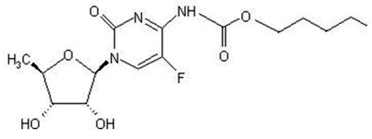
Capecitabine is een wit tot gebroken wit kristallijn poeder met een oplosbaarheid in water van 26 mg/ml bij 20°C.
XELODA wordt geleverd als biconvexe, langwerpige filmomhulde tabletten voor orale toediening. Elke licht perzikkleurige tablet bevat 150 mg capecitabine en elke perzikkleurige tablet bevat 500 mg capecitabine. De inactieve ingrediënten in XELODA zijn: watervrije lactose, croscarmellosenatrium, hydroxypropylmethylcellulose, microkristallijne cellulose, magnesiumstearaat en gezuiverd water. De perzikkleurige of licht perzikkleurige filmomhulling bevat hydroxypropylmethylcellulose, talk, titaandioxide en synthetische gele en rode ijzeroxiden.
INDICATIES
Colorectale kanker
- XELODA is geïndiceerd als monotherapie voor adjuvante behandeling bij patiënten met Dukes' C colonkanker die volledige resectie van de primaire tumor hebben ondergaan, wanneer behandeling met alleen fluoropyrimidinetherapie de voorkeur heeft. XELODA 500 mg was niet-inferieur aan 5fluorouracil en leucovorine (5-FU/LV) voor ziektevrije overleving (DFS). Artsen moeten rekening houden met de resultaten van onderzoeken met combinatiechemotherapie, die een verbetering van de DFS en OS hebben aangetoond, wanneer ze XELODA 500 mg als enkelvoudig middel voorschrijven bij de adjuvante behandeling van Dukes' C-darmkanker.
- XELODA 500 mg is geïndiceerd als eerstelijnsbehandeling van patiënten met gemetastaseerd colorectaal carcinoom wanneer behandeling met alleen fluoropyrimidinetherapie de voorkeur heeft. Combinatiechemotherapie heeft een overlevingsvoordeel laten zien in vergelijking met alleen 5-FU/LV. Een overlevingsvoordeel ten opzichte van 5-FU/LV is niet aangetoond met XELODA 500 mg monotherapie. Het gebruik van XELODA in plaats van 5FU/LV in combinaties is niet voldoende onderzocht om de veiligheid of het behoud van het overlevingsvoordeel te garanderen.
Borstkanker
- XELODA in combinatie met docetaxel is geïndiceerd voor de behandeling van patiënten met gemetastaseerde borstkanker nadat eerdere antracycline-bevattende chemotherapie heeft gefaald.
- XELODA 500 mg monotherapie is ook geïndiceerd voor de behandeling van patiënten met gemetastaseerde borstkanker die resistent is tegen zowel paclitaxel als een antracycline-bevattend chemotherapieregime of resistent is tegen paclitaxel en voor wie verdere antracyclinetherapie niet geïndiceerd is (bijv. patiënten die cumulatieve doses van 400 mg/m2 doxorubicine of doxorubicine-equivalenten). Resistentie wordt gedefinieerd als progressieve ziekte tijdens de behandeling, met of zonder een initiële respons, of terugval binnen 6 maanden na voltooiing van de behandeling met een antracyclinebevattend adjuvans regime.
DOSERING EN ADMINISTRATIE
Belangrijke administratie-instructies
XELODA-tabletten moeten binnen 30 minuten na een maaltijd in hun geheel met water worden doorgeslikt. XELODA 500 mg is een cytotoxisch geneesmiddel. Volg de toepasselijke speciale procedures voor hantering en verwijdering.1 Als XELODA 500 mg tabletten moeten worden gesneden of fijngemaakt, moet dit worden gedaan door een professional die is opgeleid in het veilig omgaan met cytotoxische geneesmiddelen met gebruikmaking van geschikte apparatuur en veiligheidsprocedures. De dosis XELODA wordt berekend op basis van het lichaamsoppervlak.
Standaard startdosis
Monotherapie (gemetastaseerde colorectale kanker, adjuvante colorectale kanker, gemetastaseerde borstkanker)
De aanbevolen dosis XELODA is 1250 mg/m2 tweemaal daags oraal toegediend ('s morgens en' s avonds; equivalent aan 2500 mg/m2 totale dagelijkse dosis) gedurende 2 weken, gevolgd door een rustperiode van 1 week, gegeven als cycli van 3 weken (zie ).
Adjuvante behandeling bij patiënten met Dukes' C colonkanker wordt aanbevolen voor in totaal 6 maanden [dwz XELODA 1250 mg/m2 tweemaal daags oraal gedurende 2 weken gevolgd door een rustperiode van 1 week, gegeven als cycli van 3 weken voor een totaal van 8 cycli (24 weken)].
In combinatie met Docetaxel (gemetastaseerde borstkanker)
In combinatie met docetaxel is de aanbevolen dosis XELODA 1250 mg/m2 tweemaal daags gedurende 2 weken gevolgd door een rustperiode van 1 week, gecombineerd met docetaxel van 75 mg/m2 als een 1-uurs intraveneuze infusie om de 3 weken. Premedicatie, volgens de docetaxel-etikettering, moet worden gestart vóór toediening van docetaxel voor patiënten die de combinatie XELODA plus docetaxel krijgen. toont de totale dagelijkse dosis XELODA per lichaamsoppervlak en het aantal tabletten dat bij elke dosis moet worden ingenomen.
Richtlijnen voor dosisbeheer
Algemeen
De dosering van XELODA moet mogelijk worden geïndividualiseerd om de behandeling van de patiënt te optimaliseren. Patiënten moeten zorgvuldig worden gecontroleerd op toxiciteit en doses van XELODA 500 mg moeten zo nodig worden aangepast om tegemoet te komen aan de individuele tolerantie van de patiënt voor de behandeling [zie Klinische studies ]. Toxiciteit als gevolg van toediening van XELODA 500 mg kan worden behandeld door symptomatische behandeling, dosisonderbrekingen en aanpassing van de XELODA-dosis. Als de dosis eenmaal is verlaagd, mag deze op een later tijdstip niet meer worden verhoogd. Doses XELODA 500 mg die zijn weggelaten wegens toxiciteit worden niet vervangen of hersteld; in plaats daarvan moet de patiënt de geplande behandelingscycli hervatten.
De dosis fenytoïne en de dosis van van coumarine afgeleide anticoagulantia moet mogelijk worden verlaagd wanneer een van beide geneesmiddelen gelijktijdig met XELODA wordt toegediend (zie DRUG-INTERACTIES ].
Monotherapie (gemetastaseerde colorectale kanker, adjuvante colorectale kanker, gemetastaseerde borstkanker)
Het schema voor dosisaanpassing van XELODA zoals hieronder beschreven (zie ) wordt aanbevolen voor de behandeling van bijwerkingen.
In combinatie met Docetaxel (gemetastaseerde borstkanker)
Dosisaanpassingen van XELODA 500 mg voor toxiciteit moeten worden gemaakt zoals hierboven voor XELODA. Als aan het begin van een behandelingscyclus een uitstel van de behandeling is geïndiceerd voor XELODA 500 mg of docetaxel, moet de toediening van beide middelen worden uitgesteld totdat aan de vereisten voor het opnieuw starten van beide geneesmiddelen is voldaan.
Het schema voor dosisverlaging voor docetaxel bij gebruik in combinatie met XELODA voor de behandeling van gemetastaseerde borstkanker wordt weergegeven in Tabel 3.
Aanpassing van de startdosis bij speciale populaties
Nierfunctiestoornis
Er wordt geen aanpassing van de startdosering van XELODA 500 mg aanbevolen bij patiënten met een lichte nierfunctiestoornis (creatinineklaring = 51 tot 80 ml/min [Cockroft en Gault, zoals hieronder weergegeven]). Bij patiënten met een matige nierfunctiestoornis (creatinineklaring bij aanvang = 30 tot 50 ml/min), een dosisverlaging tot 75% van de startdosering van XELODA bij gebruik als monotherapie of in combinatie met docetaxel (van 1250 mg/m2 tot 950 mg/m2 tweemaal daags) wordt aanbevolen [zie Gebruik bij specifieke populaties en KLINISCHE FARMACOLOGIE ]. Daaropvolgende dosisaanpassing wordt aanbevolen zoals beschreven in en (afhankelijk van het regime) als een patiënt een bijwerking van graad 2 tot 4 ontwikkelt [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. De aanbevelingen voor aanpassing van de aanvangsdosis voor patiënten met een matige nierfunctiestoornis zijn van toepassing op zowel XELODA 500 mg als monotherapie als XELODA 500 mg in combinatie met docetaxel.
Cockroft- en Gault-vergelijking:
Geriatrie
Artsen dienen voorzichtig te zijn bij het controleren van de effecten van XELODA 500 mg bij ouderen. Er zijn onvoldoende gegevens beschikbaar om een doseringsadvies te geven.
HOE GELEVERD
Doseringsvormen Sterke punten
XELODA wordt geleverd als biconvexe, langwerpige filmomhulde tabletten voor orale toediening. Elke licht perzikkleurige tablet bevat 150 mg capecitabine en elke perzikkleurige tablet bevat 500 mg capecitabine.
150 mg
Kleur: licht perzik Gravure: XELODA aan de ene kant en 150 aan de andere 150 mg tabletten zijn verpakt in flessen van 60 ( NDC 0004-1100-20), afzonderlijk verpakt in een doos.
500 mg
Kleur: Perzik Gravure: XELODA 500 mg aan de ene kant en 500 aan de andere 500 mg tabletten zijn verpakt in flessen van 120 ( NDC 0004-1101-50), afzonderlijk verpakt in een doos.
Opslag en behandeling
Bewaren bij 25°C (77°F); excursies toegestaan tot 15° tot 30°C (59° tot 86°F). [Zie USP-gecontroleerde kamertemperatuur]. HOUD DICHT GESLOTEN.
XELODA is een cytotoxisch geneesmiddel. Volg de van toepassing zijnde speciale procedures voor behandeling en verwijdering.1 Al het ongebruikte product moet worden vernietigd in overeenstemming met lokale voorschriften of programma's voor het terugnemen van geneesmiddelen.
REFERENTIES
1. "OSHA-gevaarlijke medicijnen." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Gedistribueerd door: Genentech USA, Inc. Een lid van de Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Herzien: mei 2021
BIJWERKINGEN
Omdat klinische onderzoeken onder sterk uiteenlopende omstandigheden worden uitgevoerd, kunnen de bijwerkingen die in de klinische onderzoeken van een geneesmiddel zijn waargenomen niet direct worden vergeleken met de percentages in de klinische onderzoeken van een ander geneesmiddel en komen mogelijk niet overeen met de percentages die in de praktijk worden waargenomen.
Adjuvante darmkanker
toont de bijwerkingen die optreden bij ≥ 5% van de patiënten uit één fase 3-onderzoek bij patiënten met Dukes' C-darmkanker die ten minste één dosis onderzoeksmedicatie kregen en ten minste één veiligheidsbeoordeling hadden. In totaal werden 995 patiënten behandeld met 1250 mg/m2 tweemaal daags XELODA 500 mg toegediend gedurende 2 weken gevolgd door een rustperiode van 1 week, en 974 patiënten kregen 5-FU en leucovorine (20 mg/m2 leucovorine IV gevolgd door 425 mg/m2 IV bolus 5FU op dag 1-5 elke 28 dagen). De mediane duur van de behandeling was 164 dagen voor met capecitabine behandelde patiënten en 145 dagen voor met 5-FU/LV behandelde patiënten. In totaal staakten respectievelijk 112 (11%) en 73 (7%) met capecitabine en 5-FU/LV behandelde patiënten de behandeling vanwege bijwerkingen. Er vielen in totaal 18 sterfgevallen door alle oorzaken, hetzij tijdens het onderzoek of binnen 28 dagen na ontvangst van het onderzoeksgeneesmiddel: 8 (0,8%) patiënten gerandomiseerd naar XELODA 500 mg en 10 (1,0%) gerandomiseerd naar 5-FU/LV.
vertoont laboratoriumafwijkingen van graad 3/4 die optreden bij ≥1% van de patiënten uit één fase 3-onderzoek bij patiënten met Dukes' C-darmkanker die ten minste één dosis onderzoeksmedicatie kregen en ten minste één veiligheidsbeoordeling hadden.
Gemetastaseerde colorectale kanker
Monotherapie
toont de bijwerkingen die optraden bij ≥ 5% van de patiënten bij het samenvoegen van de twee fase 3-onderzoeken bij eerstelijns gemetastaseerde colorectale kanker. In totaal werden 596 patiënten met uitgezaaide colorectale kanker behandeld met 1250 mg/m2 tweemaal daags XELODA, toegediend gedurende 2 weken gevolgd door een rustperiode van 1 week, en 593 patiënten kregen 5-FU en leucovorine in het Mayo-regime (20 mg/m2 leucovorine IV gevolgd door 425 mg/m2 IV bolus 5-FU, op dag 1-5, elke 28 dagen). In de gepoolde colorectale database was de mediane duur van de behandeling 139 dagen voor met capecitabine behandelde patiënten en 140 dagen voor met 5-FU/LV behandelde patiënten. Een totaal van respectievelijk 78 (13%) en 63 (11%) met capecitabine en 5-FU/LV behandelde patiënten stopten met de behandeling vanwege bijwerkingen/bijkomende ziekte. Een totaal van 82 sterfgevallen als gevolg van alle oorzaken traden op tijdens het onderzoek of binnen 28 dagen na ontvangst van het onderzoeksgeneesmiddel: 50 (8,4%) patiënten gerandomiseerd naar XELODA 500 mg en 32 (5,4%) gerandomiseerd naar 5-FU/LV.
Borstkanker
In combinatie met Docetaxel
De volgende gegevens worden getoond voor het combinatieonderzoek met XELODA 500 mg en docetaxel bij patiënten met uitgezaaide borstkanker in en. In de XELODA en docetaxel-combinatiearm werd XELODA oraal toegediend 1250 mg/m2 tweemaal daags als intermitterende therapie (2 weken behandeling gevolgd door 1 week zonder behandeling) gedurende ten minste 6 weken en docetaxel toegediend als een 1 uur durende intraveneuze infusie op een dosis van 75 mg/m2 op de eerste dag van elke cyclus van 3 weken gedurende ten minste 6 weken. In de monotherapiearm werd docetaxel gedurende ten minste 6 weken toegediend als een 1-uurs intraveneuze infusie in een dosis van 100 mg/m2 op de eerste dag van elke cyclus van 3 weken. De gemiddelde behandelingsduur was 129 dagen in de combinatie-arm en 98 dagen in de monotherapie-arm. Een totaal van 66 patiënten (26%) in de combinatie-arm en 49 (19%) in de monotherapie-arm trokken zich terug uit het onderzoek vanwege bijwerkingen. Het percentage patiënten dat dosisverlagingen nodig had vanwege bijwerkingen was 65% in de combinatie-arm en 36% in de monotherapie-arm. Het percentage patiënten dat onderbrekingen van de behandeling nodig had vanwege bijwerkingen in de combinatie-arm was 79%. Onderbrekingen van de behandeling maakten deel uit van het dosisaanpassingsschema voor de combinatietherapie-arm, maar niet voor de met docetaxel monotherapie behandelde patiënten.
Monotherapie
De volgende gegevens worden getoond voor de studie bij borstkankerpatiënten in stadium IV die een dosis van 1250 mg/m2 tweemaal daags kregen toegediend gedurende 2 weken, gevolgd door een rustperiode van 1 week. De gemiddelde duur van de behandeling was 114 dagen. In totaal 13 van de 162 patiënten (8%) stopten met de behandeling vanwege bijwerkingen/bijkomende ziekte.
Klinisch relevante bijwerkingen bij
Klinisch relevante bijwerkingen gemeld bij
Monotherapie (gemetastaseerde colorectale kanker, adjuvante colorectale kanker, gemetastaseerde borstkanker)
Gastro-intestinaal: opgezette buik, dysfagie, proctalgie, ascites (0,1%), maagzweer (0,1%), ileus (0,3%), toxische verwijding van de darm, gastro-enteritis (0,1%)
Huid & onderhuid.: nagelaandoening (0,1%), toegenomen zweten (0,1%), fotosensitiviteitsreactie (0,1%), huidulceratie, pruritus, stralingsherinneringssyndroom (0,2%)
Algemeen: pijn op de borst (0,2%), griepachtige ziekte, opvliegers, pijn (0,1%), heesheid, prikkelbaarheid, moeilijk lopen, dorst, borstmassa, collaps, fibrose (0,1%), bloeding, oedeem, sedatie
Neurologisch: slapeloosheid, ataxie (0,5%), tremor, dysfasie, encefalopathie (0,1%), abnormale coördinatie, dysartrie, bewustzijnsverlies (0,2%), verstoord evenwicht
Metabolisme: gewichtstoename, cachexie (0,4%), hypertriglyceridemie (0,1%), hypokaliëmie, hypomagnesiëmie
Oog: conjunctivitis
Ademhaling: hoesten (0,1%), epistaxis (0,1%), astma (0,2%), bloedspuwing, ademnood (0,1%), kortademigheid
Hart: tachycardie (0,1%), bradycardie, atriale fibrillatie, ventriculaire extrasystolen, extrasystolen, myocarditis (0,1%), pericardiale effusie
infecties: laryngitis (1,0%), bronchitis (0,2%), pneumonie (0,2%), bronchopneumonie (0,2%), keratoconjunctivitis, sepsis (0,3%), schimmelinfecties (inclusief candidiasis) (0,2%)
Musculoskeletaal: spierpijn, botpijn (0,1%), artritis (0,1%), spierzwakte
Bloed & Lymfe: leukopenie (0,2%), stollingsstoornis (0,1%), beenmergdepressie (0,1%), idiopathische trombocytopenie purpura (1,0%), pancytopenie (0,1%)
Vasculair: hypotensie (0,2%), hypertensie (0,1%), lymfoedeem (0,1%), longembolie (0,2%), cerebrovasculair accident (0,1%)
Psychiatrisch: depressie, verwardheid (0,1%)
nier: nierfunctiestoornis (0,6%)
Oor: hoogtevrees
Lever- en gal: leverfibrose (0,1%), hepatitis (0,1%), cholestatische hepatitis (0,1%), abnormale leverfunctietesten
Immuunsysteem: overgevoeligheid voor geneesmiddelen (0,1%)
XELODA 500 mg in combinatie met docetaxel (gemetastaseerde borstkanker)
Gastro-intestinaal: ileus (0,4%), necrotiserende enterocolitis (0,4%), slokdarmzweer (0,4%), hemorragische diarree (0,8%)
Neurologisch: ataxie (0,4%), syncope (1,2%), smaakverlies (0,8%), polyneuropathie (0,4%), migraine (0,4%)
Hart: supraventriculaire tachycardie (0,4%)
Infectie: neutropenische sepsis (2,4%), sepsis (0,4%), bronchopneumonie (0,4%) Bloed &
lymfatisch: agranulocytose (0,4%), protrombine verlaagd (0,4%)
Vasculair: hypotensie (1,2%), veneuze flebitis en tromboflebitis (0,4%), orthostatische hypotensie (0,8%)
nier: nierfalen (0,4%)
Lever- en gal: geelzucht (0,4%), abnormale leverfunctietesten (0,4%), leverfalen (0,4%), levercoma (0,4%), hepatotoxiciteit (0,4%)
Immuunsysteem: overgevoeligheid (1,2%)
Postmarketingervaring
De volgende bijwerkingen zijn waargenomen in de postmarketingsetting: angio-oedeem, leverfalen, traankanaalstenose, acuut nierfalen secundair aan uitdroging inclusief fatale afloop [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ], cutane lupus erythematosus, cornea-aandoeningen waaronder keratitis, toxische leuko-encefalopathie, ernstige huidreacties zoals Stevens-Johnson-syndroom en toxische epidermale necrolyse (TEN) [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ], aanhoudend of ernstig hand-en-voetsyndroom kan uiteindelijk leiden tot verlies van vingerafdrukken [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
In gevallen van blootstelling aan fijngemaakte XELODA 500 mg tabletten zijn de volgende bijwerkingen gemeld: oogirritatie en zwelling, huiduitslag, diarree, paresthesie, hoofdpijn, maagirritatie, braken en misselijkheid.
DRUG-INTERACTIES
Geneesmiddel-geneesmiddelinteracties
anticoagulantia
Veranderde stollingsparameters en/of bloedingen zijn gemeld bij patiënten die XELODA 500 mg gelijktijdig innamen met van coumarine afgeleide anticoagulantia zoals warfarine en fenprocoumon [zie DOOS WAARSCHUWING: ]. Deze voorvallen traden op binnen enkele dagen en tot enkele maanden na het starten van de behandeling met XELODA 500 mg en, in enkele gevallen, binnen 1 maand na het stoppen met XELODA. Deze voorvallen traden op bij patiënten met en zonder levermetastasen. In een geneesmiddelinteractiestudie met toediening van een enkelvoudige dosis warfarine was er een significante toename van de gemiddelde AUC van S-warfarine [zie KLINISCHE FARMACOLOGIE ]. De maximaal waargenomen INR-waarde steeg met 91%. Deze interactie is waarschijnlijk te wijten aan een remming van cytochroom P450 2C9 door capecitabine en/of zijn metabolieten.
fenytoïne
Het niveau van fenytoïne moet zorgvuldig worden gecontroleerd bij patiënten die XELODA 500 mg gebruiken en het kan nodig zijn de dosis fenytoïne te verlagen [zie DOSERING EN ADMINISTRATIE ]. Postmarketingrapporten geven aan dat sommige patiënten die XELODA 500 mg en fenytoïne kregen, toxiciteit vertoonden die gepaard ging met verhoogde fenytoïnespiegels. Er zijn geen formele onderzoeken naar geneesmiddelinteracties met fenytoïne uitgevoerd, maar er wordt aangenomen dat het interactiemechanisme remming van het CYP2C9-iso-enzym door capecitabine en/of zijn metabolieten is.
Leucovorine
De concentratie van 5-fluorouracil is verhoogd en de toxiciteit ervan kan worden versterkt door leucovorine. Sterfgevallen door ernstige enterocolitis, diarree en uitdroging zijn gemeld bij oudere patiënten die wekelijks leucovorine en fluorouracil kregen.
CYP2C9-substraten
Behalve warfarine zijn er geen formele geneesmiddelinteractiestudies uitgevoerd tussen XELODA en andere CYP2C9-substraten. Voorzichtigheid is geboden wanneer XELODA gelijktijdig wordt toegediend met CYP2C9-substraten.
Allopurinol
Gelijktijdig gebruik met allopurinol kan de concentratie van de actieve metabolieten van capecitabine verlagen (zie: KLINISCHE FARMACOLOGIE ], wat de werkzaamheid van XELODA kan verminderen. Vermijd het gebruik van allopurinol tijdens de behandeling met XELODA.
Geneesmiddel-voedsel interactie
Voedsel bleek zowel de snelheid als de mate van absorptie van capecitabine te verminderen [zie: KLINISCHE FARMACOLOGIE ]. In alle klinische onderzoeken kregen patiënten de instructie om XELODA 500 mg binnen 30 minuten na een maaltijd toe te dienen. Het wordt aanbevolen om XELODA met voedsel toe te dienen [zie: DOSERING EN ADMINISTRATIE ].
WAARSCHUWINGEN
Inbegrepen als onderdeel van de "PREVENTIEVE MAATREGELEN" Sectie
PREVENTIEVE MAATREGELEN
Coagulopathie
Patiënten die gelijktijdig met capecitabine en orale cumarine-afgeleide anticoagulantiatherapie krijgen, moeten hun anticoagulantiarespons (INR of protrombinetijd) nauwlettend en met grote frequentie laten controleren en de dosis anticoagulantia moet dienovereenkomstig worden aangepast [zie DOOS WAARSCHUWING: en DRUG-INTERACTIES ].
Diarree
XELODA 500 mg kan diarree veroorzaken, soms ernstig. Patiënten met ernstige diarree moeten zorgvuldig worden gecontroleerd en moeten vocht- en elektrolytenvervanging krijgen als ze uitgedroogd raken. Bij 875 patiënten met ofwel gemetastaseerde borst- of colorectale kanker die XELODA monotherapie kregen, was de mediane tijd tot het eerste optreden van graad 2 tot 4 diarree 34 dagen (bereik van 1 tot 369 dagen). De mediane duur van graad 3 tot 4 diarree was 5 dagen. National Cancer Institute of Canada (NCIC) graad 2 diarree wordt gedefinieerd als een toename van 4 tot 6 ontlastingen/dag of nachtelijke ontlasting, graad 3 diarree als een toename van 7 tot 9 ontlastingen/dag of incontinentie en malabsorptie, en graad 4 diarree als een toename van ≥10 ontlasting/dag of ernstig bloederige diarree of de behoefte aan parenterale ondersteuning. Als diarree van graad 2, 3 of 4 optreedt, moet de toediening van XELODA onmiddellijk worden onderbroken totdat de diarree verdwijnt of in intensiteit afneemt tot graad 1 [zie DOSERING EN ADMINISTRATIE ]. Standaardbehandelingen tegen diarree (bijv. loperamide) worden aanbevolen.
Necrotiserende enterocolitis (typhlitis) is gemeld.
Cardiotoxiciteit
De cardiotoxiciteit die is waargenomen met XELODA 500 mg omvat myocardinfarct/ischemie, angina, ritmestoornissen, hartstilstand, hartfalen, plotselinge dood, elektrocardiografische veranderingen en cardiomyopathie. Deze bijwerkingen kunnen vaker voorkomen bij patiënten met een voorgeschiedenis van coronaire hartziekte.
Dihydropyrimidine-dehydrogenasedeficiëntie
Op basis van postmarketingrapporten lopen patiënten met bepaalde homozygote of bepaalde samengestelde heterozygote mutaties in het DPD-gen die resulteren in volledige of bijna volledige afwezigheid van DPD-activiteit een verhoogd risico op acute vroege toxiciteit en ernstige, levensbedreigende of fatale bijwerkingen. reacties veroorzaakt door XELODA (bijv. mucositis, diarree, neutropenie en neurotoxiciteit). Patiënten met gedeeltelijke DPD-activiteit kunnen ook een verhoogd risico hebben op ernstige, levensbedreigende of fatale bijwerkingen veroorzaakt door XELODA.
Houd XELODA 500 mg niet of definitief stop op basis van klinische beoordeling van het begin, de duur en de ernst van de waargenomen toxiciteiten bij patiënten met tekenen van acute vroege of ongewoon ernstige toxiciteit, wat kan wijzen op een bijna volledige of totale afwezigheid van DPD-activiteit. Geen enkele dosis XELODA is veilig gebleken voor patiënten met volledige afwezigheid van DPD-activiteit. Er zijn onvoldoende gegevens om een specifieke dosis aan te bevelen bij patiënten met gedeeltelijke DPD-activiteit zoals gemeten met een specifieke test.
Uitdroging en nierfalen
Uitdroging is waargenomen en kan acuut nierfalen veroorzaken, dat fataal kan zijn. Patiënten met een reeds bestaande verminderde nierfunctie of die XELODA gelijktijdig met bekende nefrotoxische middelen krijgen, lopen een groter risico. Patiënten met anorexia, asthenie, misselijkheid, braken of diarree kunnen snel uitgedroogd raken. Controleer patiënten wanneer XELODA 500 mg wordt toegediend om uitdroging vanaf het begin te voorkomen en te corrigeren. Als uitdroging van graad 2 (of hoger) optreedt, moet de behandeling met XELODA onmiddellijk worden onderbroken en moet de uitdroging worden gecorrigeerd. De behandeling mag niet worden hervat totdat de patiënt is gerehydrateerd en eventuele precipiterende oorzaken zijn gecorrigeerd of onder controle gebracht. Indien nodig dienen dosisaanpassingen te worden toegepast voor de precipiterende bijwerking [zie: DOSERING EN ADMINISTRATIE ].
Patiënten met een matige nierfunctiestoornis bij aanvang hebben dosisverlaging nodig [zie: DOSERING EN ADMINISTRATIE ]. Patiënten met lichte en matige nierinsufficiëntie bij aanvang moeten zorgvuldig worden gecontroleerd op bijwerkingen. Een onmiddellijke onderbreking van de behandeling met daaropvolgende dosisaanpassingen wordt aanbevolen als een patiënt een bijwerking van graad 2 tot 4 ontwikkelt, zoals beschreven in [zie DOSERING EN ADMINISTRATIE , Gebruik bij specifieke populaties , en KLINISCHE FARMACOLOGIE ].
Embryo-foetale toxiciteit
Op basis van bevindingen uit reproductiestudies bij dieren en het werkingsmechanisme kan XELODA schade aan de foetus veroorzaken wanneer het wordt toegediend aan een zwangere vrouw [zie KLINISCHE FARMACOLOGIE ]. Beperkte beschikbare gegevens zijn niet voldoende om het gebruik van XELODA 500 mg bij zwangere vrouwen te informeren. In reproductiestudies bij dieren veroorzaakte toediening van capecitabine aan drachtige dieren tijdens de periode van organogenese embryoletaliteit en teratogeniteit bij muizen en embryoletaliteit bij apen bij 0,2 en 0,6 maal de blootstelling (AUC) bij patiënten die respectievelijk de aanbevolen dosis kregen [zie Gebruik bij specifieke populaties ]. Breng zwangere vrouwen op de hoogte van het potentiële risico voor een foetus. Adviseer vrouwen in de vruchtbare leeftijd om effectieve anticonceptie te gebruiken tijdens de behandeling en gedurende 6 maanden na de laatste dosis XELODA (zie Gebruik bij specifieke populaties ].
Mucocutane en dermatologische toxiciteit
Ernstige mucocutane reacties, sommige met fatale afloop, zoals Stevens-Johnson-syndroom en toxische epidermale necrolyse (TEN) kunnen optreden bij patiënten die met XELODA worden behandeld [zie ONGEWENSTE REACTIES ]. XELODA 500 mg dient permanent te worden gestaakt bij patiënten die een ernstige mucocutane reactie ervaren die mogelijk te wijten is aan behandeling met XELODA.
Hand-en-voetsyndroom (palmair-plantaire erythrodysesthesie of door chemotherapie geïnduceerd acral erytheem) is een cutane toxiciteit. De mediane tijd tot aanvang was 79 dagen (bereik van 11 tot 360 dagen) met een ernstbereik van graad 1 tot 3 voor patiënten die XELODA 500 mg monotherapie kregen in de gemetastaseerde setting. Graad 1 wordt gekenmerkt door een van de volgende symptomen: gevoelloosheid, dysesthesie/paresthesie, tintelingen, pijnloze zwelling of erytheem van de handen en/of voeten en/of ongemak dat de normale activiteiten niet verstoort. Graad 2 hand-en-voetsyndroom wordt gedefinieerd als pijnlijk erytheem en zwelling van de handen en/of voeten en/of ongemak die de dagelijkse activiteiten van de patiënt beïnvloeden. Graad 3 hand-en-voetsyndroom wordt gedefinieerd als vochtige desquamatie, ulceratie, blaarvorming of ernstige pijn aan handen en/of voeten en/of ernstig ongemak waardoor de patiënt niet in staat is te werken of activiteiten van het dagelijks leven uit te voeren. Aanhoudend of ernstig hand-en-voetsyndroom (graad 2 en hoger) kan uiteindelijk leiden tot verlies van vingerafdrukken, wat de identificatie van de patiënt kan beïnvloeden. Als graad 2 of 3 hand-en-voetsyndroom optreedt, moet de toediening van XELODA worden onderbroken totdat het voorval is verdwenen of in intensiteit is afgenomen tot graad 1. Na graad 3 hand-en-voetsyndroom moeten de volgende doses XELODA 500 mg worden verlaagd [ zien DOSERING EN ADMINISTRATIE ].
Hyperbilirubinemie
Bij 875 patiënten met ofwel gemetastaseerde borst- of colorectale kanker die ten minste één dosis XELODA 1250 mg/m2 tweemaal daags als monotherapie kregen gedurende 2 weken gevolgd door een rustperiode van 1 week, trad graad 3 (1,5-3 x ULN) hyperbilirubinemie op in 15,2% (n=133) van de patiënten en graad 4 (>3 x ULN) hyperbilirubinemie trad op bij 3,9% (n=34) van de patiënten. Van de 566 patiënten met levermetastasen bij baseline en 309 patiënten zonder levermetastasen bij baseline, trad hyperbilirubinemie van graad 3 of 4 op bij respectievelijk 22,8% en 12,3%. Van de 167 patiënten met hyperbilirubinemie graad 3 of 4 had 18,6% (n=31) ook verhogingen na baseline (graad 1 tot 4, zonder verhogingen bij baseline) van alkalische fosfatase en had 27,5% (n=46) verhogingen van transaminasen na baseline op op elk moment (niet noodzakelijk gelijktijdig). De meerderheid van deze patiënten, 64,5% (n=20) en 71,7% (n=33), had bij baseline levermetastasen. Bovendien had 57,5% (n=96) en 35,3% (n=59) van de 167 patiënten verhogingen (graad 1 tot 4) bij zowel prebaseline als postbaseline in respectievelijk alkalische fosfatase of transaminasen. Slechts 7,8% (n=13) en 3,0% (n=5) had graad 3 of 4 verhogingen van alkalische fosfatase of transaminasen.
Bij de 596 patiënten die werden behandeld met XELODA 500 mg als eerstelijnstherapie voor gemetastaseerde colorectale kanker, was de incidentie van graad 3 of 4 hyperbilirubinemie vergelijkbaar met de algemene veiligheidsdatabase van XELODA 500 mg als monotherapie. De mediane tijd tot het optreden van hyperbilirubinemie graad 3 of 4 bij de populatie met colorectale kanker was 64 dagen en de mediane totale bilirubine nam toe van 8 m/l bij baseline tot 13 m/l tijdens behandeling met XELODA. Van de 136 patiënten met colorectale kanker met graad 3 of 4 hyperbilirubinemie, hadden 49 patiënten graad 3 of 4 hyperbilirubinemie als laatste gemeten waarde, van wie 46 levermetastasen hadden bij baseline.
Bij 251 patiënten met uitgezaaide borstkanker die een combinatie van XELODA 500 mg en docetaxel kregen, trad graad 3 (1,5 tot 3 x ULN) hyperbilirubinemie op bij 7% (n=17) en graad 4 (>3 x ULN) hyperbilirubinemie trad op bij 2% (n=5).
Als geneesmiddelgerelateerde graad 3 tot 4 verhogingen van bilirubine optreden, moet de toediening van XELODA 500 mg onmiddellijk worden onderbroken totdat de hyperbilirubinemie is afgenomen tot ≤ 3,0 X ULN (zie aanbevolen dosisaanpassingen onder DOSERING EN ADMINISTRATIE ].
hematologisch
Bij 875 patiënten met ofwel gemetastaseerde borst- of colorectale kanker die een dosis van 1250 mg/m2 kregen, tweemaal daags toegediend als monotherapie gedurende 2 weken, gevolgd door een rustperiode van 1 week, hadden 3,2%, 1,7% en 2,4% van de patiënten graad 3 of 4 respectievelijk neutropenie, trombocytopenie of afname van hemoglobine. Van 251 patiënten met uitgezaaide borstkanker die een dosis XELODA kregen in combinatie met docetaxel, had 68% graad 3 of 4 neutropenie, 2,8% had graad 3 of 4 trombocytopenie en 9,6% had graad 3 of 4 anemie.
Patiënten met baseline neutrofielentellingen van
Geriatrische patiënten
Patiënten ≥80 jaar oud kunnen een grotere incidentie van graad 3 of 4 bijwerkingen ervaren. Bij 875 patiënten met ofwel gemetastaseerde borst- of colorectale kanker die XELODA 500 mg monotherapie kregen, kreeg 62% van de 21 patiënten ≥80 jaar die werden behandeld met XELODA 500 mg een behandelingsgerelateerde bijwerking van graad 3 of 4: diarree bij 6 (28,6%) , misselijkheid bij 3 (14,3%), hand-en-voetsyndroom bij 3 (14,3%) en braken bij 2 (9,5%) patiënten. Van de 10 patiënten van 70 jaar en ouder (er waren geen patiënten >80 jaar) die werden behandeld met XELODA 500 mg in combinatie met docetaxel, kreeg 30% (3 van de 10) van de patiënten graad 3 of 4 diarree en stomatitis, en 40 % (4 van de 10) ervaren graad 3 hand-en-voetsyndroom.
Van de 67 patiënten ≥60 jaar die XELODA in combinatie met docetaxel kregen, was de incidentie van behandelingsgerelateerde bijwerkingen graad 3 of 4, behandelingsgerelateerde ernstige bijwerkingen, stopzettingen vanwege bijwerkingen, stopzetting van de behandeling vanwege bijwerkingen en behandeling stopzettingen binnen de eerste twee behandelingscycli waren hoger dan in de patiëntengroep
Bij 995 patiënten die XELODA kregen als adjuvante therapie voor Dukes' C colonkanker na resectie van de primaire tumor, kreeg 41% van de 398 patiënten ≥65 jaar die met XELODA werden behandeld een behandelingsgerelateerde bijwerking van graad 3 of 4: -voetsyndroom bij 75 (18,8%), diarree bij 52 (13,1%), stomatitis bij 12 (3,0%), neutropenie/granulocytopenie bij 11 (2,8%), braken bij 6 (1,5%) en misselijkheid bij 5 (1,3). %) patiënten. Bij patiënten ≥ 65 jaar (alle gerandomiseerde populaties; capecitabine 188 patiënten, 5-FU/LV 208 patiënten) die werden behandeld voor Dukes' C coloncarcinoom na resectie van de primaire tumor, werden de hazard ratio's voor ziektevrije overleving en algehele overleving voor XELODA vergeleken met 5-FU/LV waren respectievelijk 1,01 (95% BI 0,80 – 1,27) en 1,04 (95% CI 0,79 – 1,37).
Leverinsufficiëntie
Patiënten met een lichte tot matige leverfunctiestoornis als gevolg van levermetastasen moeten zorgvuldig worden gecontroleerd wanneer XELODA wordt toegediend. Het effect van ernstige leverdisfunctie op de beschikbaarheid van XELODA is niet bekend [zie: Gebruik bij specifieke populaties en KLINISCHE FARMACOLOGIE ].
Combinatie met andere medicijnen
Het gebruik van XELODA in combinatie met irinotecan is niet voldoende onderzocht.
Informatie over patiëntbegeleiding
Adviseer de patiënt om de door de FDA goedgekeurde patiëntetikettering te lezen ( PATIËNT INFORMATIE ).
Diarree
Informeer patiënten die diarree van graad 2 (een toename van 4 tot 6 ontlasting/dag of nachtelijke ontlasting) of meer ervaren of die ernstige bloederige diarree hebben met ernstige buikpijn en koorts, te stoppen met het gebruik van XELODA. Adviseer patiënten over het gebruik van antidiarreebehandelingen (bijv. loperamide) om diarree te behandelen [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Cardiotoxiciteit
Adviseer patiënten over het risico op cardiotoxiciteit en om onmiddellijk contact op te nemen met hun zorgverlener of om naar een eerstehulpafdeling te gaan voor nieuwe pijn op de borst, kortademigheid, duizeligheid of een licht gevoel in het hoofd [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Dihydropyrimidine-dehydrogenasedeficiëntie
Adviseer patiënten om hun zorgverlener op de hoogte te stellen als ze een bekende DPD-tekort hebben. Adviseer patiënten als ze volledige of bijna volledige afwezigheid van DPD-activiteit hebben, ze een verhoogd risico lopen op acute vroege toxiciteit en ernstige, levensbedreigende of fatale bijwerkingen veroorzaakt door XELODA (bijv. mucositis, diarree, neutropenie en neurotoxiciteit) [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Uitdroging en nierfalen
Instrueer patiënten die uitdroging van graad 2 of hoger ervaren (infuusvloeistoffen WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Belangrijke administratie-instructies
Adviseer patiënten om XELODA 500 mg tabletten in hun geheel door te slikken met water binnen 30 minuten na een maaltijd. Adviseer patiënten en zorgverleners om XELODA-tabletten niet fijn te maken of te snijden. Adviseer patiënten als ze XELODA-tabletten niet in hun geheel kunnen doorslikken, om hun zorgverlener te informeren [zie: DOSERING EN ADMINISTRATIE ].
Overgevoeligheid en angio-oedeem
Adviseer patiënten dat XELODA ernstige overgevoeligheidsreacties en angio-oedeem kan veroorzaken. Adviseer patiënten met een bekende overgevoeligheid voor capecitabine of 5-fluorouracil om hun zorgverlener te informeren [zie: CONTRA-INDICATIES ]. Instrueer patiënten die overgevoeligheidsreacties of mucocutane symptomen ontwikkelen (bijv. urticaria, huiduitslag, erytheem, pruritus of zwelling van het gezicht, lippen, tong of keel waardoor slikken of ademen bemoeilijkt) te stoppen met het gebruik van XELODA en onmiddellijk contact op te nemen met hun zorgverlener of om naar de eerste hulp te gaan. [zien ONGEWENSTE REACTIES ].
Misselijkheid
Instrueer patiënten die graad 2 misselijkheid ervaren (de voedselinname aanzienlijk verminderd maar in staat om met tussenpozen te eten) of meer om onmiddellijk te stoppen met het gebruik van XELODA 500 mg en om contact op te nemen met hun zorgverlener voor de behandeling van misselijkheid [zie ONGEWENSTE REACTIES ].

Braken
Instrueer patiënten die graad 2 braken ervaren (2 tot 5 episodes in een periode van 24 uur) of meer om onmiddellijk te stoppen met het gebruik van XELODA en om contact op te nemen met hun zorgverlener voor de behandeling van braken [zie ONGEWENSTE REACTIES ].
Hand-en-voetsyndroom
Instrueer patiënten met graad 2 hand-en-voetsyndroom (pijnlijk erytheem en zwelling van de handen en/of voeten en/of ongemak dat de dagelijkse activiteiten van de patiënt beïnvloedt) of meer om onmiddellijk te stoppen met het gebruik van XELODA 500 mg en contact op te nemen met hun zorgverlener . Informeer patiënten dat het starten van een symptomatische behandeling wordt aanbevolen en dat het hand-en-voetsyndroom kan leiden tot verlies van vingerafdrukken, wat van invloed kan zijn op persoonlijke identificatie [zie ONGEWENSTE REACTIES ].
stomatitis
Informeer patiënten die graad 2 stomatitis (pijnlijk erytheem, oedeem of zweren in de mond of tong, maar in staat om te eten) of meer ervaren om onmiddellijk te stoppen met het gebruik van XELODA en contact op te nemen met hun zorgverlener [zie ONGEWENSTE REACTIES ].
Koorts en neutropenie
Informeer patiënten die koorts krijgen van 100,5°F of hoger of ander bewijs van mogelijke infectie om contact op te nemen met hun zorgverlener [zie ONGEWENSTE REACTIES ].
Embryo-foetale toxiciteit
Adviseer vrouwen met reproductief vermogen over het mogelijke risico voor een foetus en om effectieve anticonceptie te gebruiken tijdens de behandeling met XELODA 500 mg en gedurende 6 maanden na de laatste dosis. Adviseer vrouwen om hun zorgverlener op de hoogte te stellen van een bekende of vermoede zwangerschap [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , Gebruik bij specifieke populaties ].
Adviseer mannelijke patiënten met vrouwelijke partners in de vruchtbare leeftijd om effectieve anticonceptie te gebruiken tijdens de behandeling met XELODA 500 mg en gedurende 3 maanden na de laatste dosis [zie Gebruik bij specifieke populaties ].
Borstvoeding
Adviseer vrouwen om geen borstvoeding te geven tijdens de behandeling met XELODA 500 mg en gedurende 2 weken na de laatste dosis [zie Gebruik bij specifieke populaties ].
Niet-klinische toxicologie
Carcinogenese, mutagenese, verminderde vruchtbaarheid
Adequate onderzoeken naar het carcinogene potentieel van capecitabine zijn niet uitgevoerd. Capecitabine was in vitro niet mutageen voor bacteriën (Ames-test) of zoogdiercellen (Chinese hamster V79/HPRT-genmutatietest). Capecitabine was in vitro clastogeen voor humane perifere bloedlymfocyten, maar niet clastogeen in vivo voor muisbeenmerg (micronucleustest). Fluorouracil veroorzaakt mutaties in bacteriën en gisten. Fluorouracil veroorzaakt ook chromosomale afwijkingen in de micronucleustest bij muizen in vivo.
In onderzoeken naar vruchtbaarheid en algemene reproductieprestaties bij vrouwelijke muizen, verstoorden orale doses capecitabine van 760 mg/kg/dag (ongeveer 2300 mg/m2/dag) de oestrus en veroorzaakten ze bijgevolg een afname van de vruchtbaarheid. Bij muizen die zwanger werden, overleefden geen enkele foetus deze dosis. De stoornis in de oestrus was omkeerbaar. Bij mannen veroorzaakte deze dosis degeneratieve veranderingen in de testikels, waaronder een afname van het aantal spermatocyten en spermatiden. In afzonderlijke farmacokinetische onderzoeken produceerde deze dosis bij muizen 5'-DFUR AUC-waarden van ongeveer 0,7 maal de overeenkomstige waarden bij patiënten die de aanbevolen dagelijkse dosis kregen.
Gebruik bij specifieke populaties
Zwangerschap
Risico Samenvatting
Gebaseerd op bevindingen in reproductiestudies bij dieren en het werkingsmechanisme, kan XELODA 500 mg schade aan de foetus veroorzaken wanneer het wordt toegediend aan een zwangere vrouw [zie KLINISCHE FARMACOLOGIE ]. Beperkte beschikbare gegevens bij de mens zijn niet voldoende om het geneesmiddelgerelateerde risico tijdens de zwangerschap te informeren. In reproductieonderzoeken bij dieren veroorzaakte toediening van capecitabine aan drachtige dieren tijdens de periode van organogenese embryoletaliteit en teratogeniteit bij muizen en embryoletaliteit bij apen bij 0,2 en 0,6 keer de blootstelling (AUC) bij patiënten die respectievelijk de aanbevolen dosis kregen [zie Gegevens ]. Breng zwangere vrouwen op de hoogte van het potentiële risico voor een foetus.
Het geschatte achtergrondrisico van ernstige geboorteafwijkingen en miskraam voor de aangegeven populatie is niet bekend. In de algemene bevolking van de VS is het geschatte achtergrondrisico van ernstige geboorteafwijkingen en miskraam bij klinisch erkende zwangerschappen respectievelijk 2-4% en 15-20%.
Gegevens
Dierlijke gegevens
Orale toediening van capecitabine aan zwangere muizen tijdens de periode van organogenese in een dosis van 198 mg/kg/dag veroorzaakte misvormingen en embryonale letaliteit. In afzonderlijke farmacokinetische onderzoeken produceerde deze dosis bij muizen 5'-DFUR AUC-waarden die ongeveer 0,2 maal de AUC-waarden waren bij patiënten die de aanbevolen dagelijkse dosis kregen. Misvormingen bij muizen omvatten gespleten gehemelte, anoftalmie, microftalmie, oligodactylie, polydactylie, syndactylie, kinky staart en verwijding van cerebrale ventrikels. Orale toediening van capecitabine aan drachtige apen tijdens de periode van organogenese in een dosis van 90 mg/kg/dag veroorzaakte foetale letaliteit. Deze dosis produceerde 5'-DFUR AUC-waarden die ongeveer 0,6 maal de AUC-waarden waren bij patiënten die de aanbevolen dagelijkse dosis kregen.
Borstvoeding
Risico Samenvatting
Er is geen informatie over de aanwezigheid van capecitabine in moedermelk, of over de effecten ervan op de melkproductie of de zuigeling die borstvoeding krijgt. Metabolieten van capecitabine waren aanwezig in de melk van zogende muizen [zie Gegevens ]. Vanwege de mogelijkheid van ernstige bijwerkingen van blootstelling aan capecitabine bij zuigelingen die borstvoeding krijgen, adviseren vrouwen om geen borstvoeding te geven tijdens de behandeling met XELODA 500 mg en gedurende 2 weken na de laatste dosis.
Gegevens
Zogende muizen die een enkele orale dosis capecitabine kregen, scheidden significante hoeveelheden capecitabinemetabolieten uit in de melk.
Vrouwtjes en mannetjes met reproductief potentieel
Zwangerschapstesten
Zwangerschapstests worden aanbevolen voor vrouwen in de vruchtbare leeftijd voordat met XELODA wordt begonnen.
anticonceptie
vrouwen
XELODA 500 mg kan schade aan de foetus veroorzaken wanneer het wordt toegediend aan een zwangere vrouw [zie Zwangerschap ]. Adviseer vrouwen in de vruchtbare leeftijd om effectieve anticonceptie te gebruiken tijdens de behandeling en gedurende 6 maanden na de laatste dosis XELODA.
mannen
Adviseer op basis van genetische toxiciteitsbevindingen mannelijke patiënten met vrouwelijke partners van voortplantingsvermogen om effectieve anticonceptie te gebruiken tijdens de behandeling en gedurende 3 maanden na de laatste dosis XELODA (zie Niet-klinische toxicologie ].
Onvruchtbaarheid
Op basis van dierstudies kan XELODA de vruchtbaarheid bij vrouwen en mannen met voortplantingsvermogen verminderen [zie: Niet-klinische toxicologie ].
Pediatrisch gebruik
De veiligheid en werkzaamheid van XELODA bij pediatrische patiënten zijn niet vastgesteld. Er werd geen klinisch voordeel aangetoond in twee eenarmige onderzoeken bij pediatrische patiënten met nieuw gediagnosticeerde hersenstamgliomen en hooggradige gliomen. In beide onderzoeken kregen pediatrische patiënten een experimentele pediatrische formulering van capecitabine gelijktijdig met en na voltooiing van de bestralingstherapie (totale dosis van 5580 cGy in 180 cGy-fracties). De relatieve biologische beschikbaarheid van de onderzoeksformulering met XELODA 500 mg was vergelijkbaar.
De eerste studie werd uitgevoerd bij 22 pediatrische patiënten (mediane leeftijd 8 jaar, bereik 5-17 jaar) met nieuw gediagnosticeerde niet-gedissemineerde intrinsieke diffuse hersenstamgliomen en hooggradige gliomen. In het dosisbepalingsgedeelte van de studie kregen patiënten capecitabine met gelijktijdige bestralingstherapie in doses variërend van 500 mg/m2 tot 850 mg/m2 elke 12 uur gedurende maximaal 9 weken. Na een pauze van 2 weken kregen de patiënten elke 12 uur 1250 mg/m2 capecitabine op dag 1-14 van een cyclus van 21 dagen gedurende maximaal 3 cycli. De maximaal getolereerde dosis (MTD) van capecitabine die gelijktijdig met radiotherapie werd toegediend, was 650 mg/m2 elke 12 uur. De belangrijkste dosisbeperkende toxiciteiten waren palmoplantaire erythrodysesthesie en verhoging van alanineaminotransferase (ALT).
De tweede studie werd uitgevoerd bij 34 extra pediatrische patiënten met nieuw gediagnosticeerde niet-gedissemineerde intrinsieke diffuse hersenstamgliomen (mediane leeftijd 7 jaar, bereik 3-16 jaar) en 10 pediatrische patiënten die de MTD van capecitabine kregen in de dosisbepalingsstudie en de geschiktheidscriteria voor deze proef. Alle patiënten kregen gedurende maximaal 9 weken elke 12 uur 650 mg/m2 capecitabine met gelijktijdige bestralingstherapie. Na een pauze van 2 weken kregen de patiënten elke 12 uur 1250 mg/m2 capecitabine op dag 1-14 van een cyclus van 21 dagen gedurende maximaal 3 cycli.
Er was geen verbetering van de progressievrije overleving na een jaar en de algehele overleving na een jaar bij pediatrische patiënten met nieuw gediagnosticeerde intrinsieke hersenstamgliomen die capecitabine kregen in vergelijking met een vergelijkbare populatie van pediatrische patiënten die deelnamen aan andere klinische onderzoeken.
Het bijwerkingenprofiel van capecitabine kwam overeen met het bekende bijwerkingenprofiel bij volwassenen, met uitzondering van laboratoriumafwijkingen die vaker voorkwamen bij pediatrische patiënten. De meest gemelde laboratoriumafwijkingen (incidentie per patiënt ≥ 40%) waren verhoogde ALT (75%), lymfocytopenie (73%), leukopenie (73%), hypokaliëmie (68%), trombocytopenie (57%), hypoalbuminemie (55 %), neutropenie (50%), lage hematocriet (50%), hypocalciëmie (48%), hypofosfatemie (45%) en hyponatriëmie (45%).
Geriatrisch gebruik
Artsen moeten bijzondere aandacht besteden aan het controleren van de bijwerkingen van XELODA 500 mg bij ouderen [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Leverinsufficiëntie
Wees voorzichtig wanneer patiënten met een lichte tot matige leverfunctiestoornis als gevolg van levermetastasen worden behandeld met XELODA. Het effect van ernstige leverdisfunctie op XELODA is niet bekend [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en KLINISCHE FARMACOLOGIE ].
Nierinsufficiëntie
Patiënten met een matige (creatinineklaring = 30 tot 50 ml/min) en ernstige (creatinineklaring CONTRA-INDICATIES , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , DOSERING EN ADMINISTRATIE , en KLINISCHE FARMACOLOGIE ].
OVERDOSERING
De manifestaties van acute overdosering zijn misselijkheid, braken, diarree, gastro-intestinale irritatie en bloeding, en beenmergdepressie. De medische behandeling van overdosering moet de gebruikelijke ondersteunende medische interventies omvatten die gericht zijn op het corrigeren van de zich voordoende klinische manifestaties. Hoewel er geen klinische ervaring is gemeld met dialyse als behandeling voor een overdosis XELODA 500 mg, kan dialyse nuttig zijn bij het verlagen van de circulerende concentraties van 5'-DFUR, een metaboliet met een laag molecuulgewicht van de moederverbinding.
Enkelvoudige doses XELODA 500 mg waren niet dodelijk voor muizen, ratten en apen bij doses tot 2000 mg/kg (2,4, 4,8 en 9,6 maal de aanbevolen dagelijkse dosis voor mensen op basis van mg/m2).
CONTRA-INDICATIES
Ernstige nierfunctiestoornis
XELODA is gecontra-indiceerd bij patiënten met een ernstige nierfunctiestoornis (creatinineklaring lager dan 30 ml/min [Cockroft en Gault]) [zie Gebruik bij specifieke populaties en KLINISCHE FARMACOLOGIE ].
overgevoeligheid
XELODA 500 mg is gecontra-indiceerd bij patiënten met een bekende overgevoeligheid voor capecitabine of voor een van de bestanddelen ervan. XELODA is gecontra-indiceerd bij patiënten met een bekende overgevoeligheid voor 5fluorouracil.
KLINISCHE FARMACOLOGIE
Werkingsmechanisme
Enzymen zetten capecitabine in vivo om in 5-fluorouracil (5-FU). Zowel normale als tumorcellen metaboliseren 5-FU tot 5-fluor-2'-deoxyuridinemonofosfaat (FdUMP) en 5-fluorouridinetrifosfaat (FUTP). Deze metabolieten veroorzaken celbeschadiging door twee verschillende mechanismen. Ten eerste binden FdUMP en de folaat-cofactor, N5-10-methyleentetrahydrofolaat, aan thymidylaatsynthase (TS) om een covalent gebonden ternair complex te vormen. Deze binding remt de vorming van thymidylaat uit 2'-deoxyuridylaat. Thymidylaat is de noodzakelijke voorloper van thymidinetrifosfaat, dat essentieel is voor de synthese van DNA, zodat een tekort aan deze verbinding de celdeling kan remmen. Ten tweede kunnen nucleaire transcriptionele enzymen per ongeluk FUTP opnemen in plaats van uridinetrifosfaat (UTP) tijdens de synthese van RNA. Deze metabolische fout kan de RNA-verwerking en eiwitsynthese verstoren.
Farmacokinetiek
Absorptie
Na orale toediening van 1255 mg/m2 tweemaal daags aan kankerpatiënten, bereikte capecitabine piekbloedspiegels in ongeveer 1,5 uur (Tmax) met piek 5-FU-spiegels die iets later optraden, na 2 uur. Voedsel verminderde zowel de snelheid als de mate van absorptie van capecitabine, waarbij de gemiddelde Cmax en AUC0-∞ daalden met respectievelijk 60% en 35%. De Cmax en AUC0-∞ van 5-FU werden ook verlaagd door voedsel met respectievelijk 43% en 21%. Voedsel vertraagde Tmax van zowel ouder als 5-FU met 1,5 uur [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , DOSERING EN ADMINISTRATIE , en Geneesmiddel-voedsel interactie ].
De farmacokinetiek van XELODA 500 mg en zijn metabolieten zijn geëvalueerd bij ongeveer 200 kankerpatiënten over een doseringsbereik van 500 tot 3500 mg/m2/dag. Binnen dit bereik was de farmacokinetiek van XELODA en zijn metaboliet, 5'-DFCR, dosisproportioneel en veranderde deze niet in de loop van de tijd. De verhogingen van de AUC's van 5'-DFUR en 5-FU waren echter meer dan evenredig met de dosisverhoging en de AUC van 5-FU was op dag 14 34% hoger dan op dag 1. De interpatiëntvariabiliteit in de Cmax en AUC van 5-FU waren meer dan 85%.
Verdeling
De plasma-eiwitbinding van capecitabine en zijn metabolieten is minder dan 60% en is niet afhankelijk van de concentratie. Capecitabine werd voornamelijk gebonden aan humaan albumine (ongeveer 35%). XELODA 500 mg heeft een laag potentieel voor farmacokinetische interacties gerelateerd aan plasma-eiwitbinding.
Bioactivatie en metabolisme
Capecitabine wordt uitgebreid enzymatisch gemetaboliseerd tot 5-FU. In de lever hydrolyseert een carboxylesterase van 60 kDa veel van de verbinding tot 5'-deoxy-5-fluorocytidine (5'-DFCR). Cytidinedeaminase, een enzym dat in de meeste weefsels wordt aangetroffen, inclusief tumoren, zet vervolgens 5'-DFCR om in 5'-DFUR. Het enzym, thymidinefosforylase (dThdPase), hydrolyseert vervolgens 5'DFUR tot het actieve geneesmiddel 5-FU. Veel weefsels door het hele lichaam brengen thymidinefosforylase tot expressie. Sommige menselijke carcinomen brengen dit enzym in hogere concentraties tot expressie dan omringende normale weefsels. Na orale toediening van XELODA 7 dagen vóór de operatie aan patiënten met colorectale kanker, was de mediane verhouding van de 5-FU-concentratie in colorectale tumoren tot aangrenzende weefsels 2,9 (bereik van 0,9 tot 8,0). Deze verhoudingen zijn niet geëvalueerd bij borstkankerpatiënten of vergeleken met 5-FU-infusie.
Metabole route van capecitabine naar 5-FU
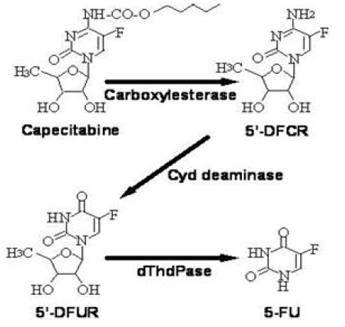
Het enzym dihydropyrimidine dehydrogenase hydrogeneert 5-FU, het product van het capecitabinemetabolisme, tot het veel minder giftige 5-fluoro-5,6-dihydro-fluorouracil (FUH2). Dihydropyrimidinase splitst de pyrimidinering om 5-fluor-ureido-propionzuur (FUPA) op te leveren. Ten slotte splitst β-ureido-propionase FUPA tot α-fluor-β-alanine (FBAL), dat in de urine wordt geklaard.
In vitro enzymatische onderzoeken met humane levermicrosomen gaven aan dat capecitabine en zijn metabolieten (5'-DFUR, 5'-DFCR, 5-FU en FBAL) het metabolisme van testsubstraten door cytochroom P450 iso-enzymen 1A2, 2A6, 3A4, 2C19, 2D6 en 2E1.
uitscheiding
Capecitabine en zijn metabolieten worden voornamelijk uitgescheiden in de urine; 95,5% van de toegediende dosis capecitabine wordt teruggevonden in de urine. Fecale excretie is minimaal (2,6%). De belangrijkste metaboliet die in de urine wordt uitgescheiden, is FBAL, dat 57% van de toegediende dosis vertegenwoordigt. Ongeveer 3% van de toegediende dosis wordt als onveranderd geneesmiddel in de urine uitgescheiden. De eliminatiehalfwaardetijd van zowel capecitabine als 5-FU was ongeveer 0,75 uur.
Effect van leeftijd, geslacht en ras op de farmacokinetiek van capecitabine
Een populatieanalyse van gepoolde gegevens van de twee grote gecontroleerde onderzoeken bij patiënten met gemetastaseerde colorectale kanker (n=505) die tweemaal daags 500 mg XELODA 1250 mg/m2 kregen toegediend, toonde aan dat geslacht (202 vrouwen en 303 mannen) en ras (455 blanke/blanke patiënten, 22 zwarte patiënten en 28 patiënten van een ander ras) hebben geen invloed op de farmacokinetiek van 5'DFUR, 5-FU en FBAL. Leeftijd heeft geen significante invloed op de farmacokinetiek van 5'-DFUR en 5-FU in het bereik van 27 tot 86 jaar. Een toename van 20% in leeftijd resulteert in een toename van 15% in AUC van FBAL [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en DOSERING EN ADMINISTRATIE ].
Na orale toediening van 825 mg/m2 capecitabine tweemaal daags gedurende 14 dagen, hadden Japanse patiënten (n=18) ongeveer 36% lagere Cmax en 24% lagere AUC voor capecitabine dan de blanke patiënten (n=22). Japanse patiënten hadden ook ongeveer 25% lagere Cmax en 34% lagere AUC voor FBAL dan de blanke patiënten. De klinische betekenis van deze verschillen is niet bekend. Er traden geen significante verschillen op in de blootstelling aan andere metabolieten (5'-DFCR, 5'-DFUR en 5FU).
Effect van leverinsufficiëntie
XELODA is geëvalueerd bij 13 patiënten met milde tot matige leverdisfunctie als gevolg van levermetastasen gedefinieerd door een samengestelde score inclusief bilirubine, ASAT/ALAT en alkalische fosfatase na een enkelvoudige dosis van 1255 mg/m2 XELODA. Zowel de AUC0-∞ als de Cmax van capecitabine namen met 60% toe bij patiënten met een leverfunctiestoornis in vergelijking met patiënten met een normale leverfunctie (n=14). De AUC0-∞ en Cmax van 5-FU werden niet beïnvloed. Bij patiënten met een lichte tot matige leverfunctiestoornis als gevolg van levermetastasen is voorzichtigheid geboden wanneer XELODA 500 mg wordt toegediend. Het effect van ernstige leverdisfunctie op XELODA 500 mg is niet bekend [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN en Gebruik in speciale populaties ].
Effect van nierinsufficiëntie
Na orale toediening van 1250 mg/m2 capecitabine tweemaal daags aan kankerpatiënten met verschillende gradaties van nierfunctiestoornis, vertoonden patiënten met matige (creatinineklaring = 30 tot 50 ml/min) en ernstige (creatinineklaring 80 ml/min). Systemische blootstelling aan 5'-DFUR was respectievelijk 42% en 71% hoger bij patiënten met een matige en ernstige nierfunctiestoornis dan bij normale patiënten. De systemische blootstelling aan capecitabine was ongeveer 25% hoger bij zowel patiënten met matige als ernstige nierfunctiestoornis [zie: DOSERING EN ADMINISTRATIE , CONTRA-INDICATIES , WAARSCHUWINGEN EN VOORZORGSMAATREGELEN , en Gebruik in speciale populaties ].
Effect van capecitabine op de farmacokinetiek van warfarine
Bij vier patiënten met kanker verhoogde chronische toediening van capecitabine (1250 mg/m2 tweemaal daags) met een enkelvoudige dosis van 20 mg warfarine de gemiddelde AUC van S-warfarine met 57% en verminderde de klaring met 37%. Op baseline gecorrigeerde AUC van INR bij deze 4 patiënten nam toe met een factor 2,8 en de maximaal waargenomen gemiddelde INR-waarde was verhoogd met 91% [zie DOOS WAARSCHUWING: en DRUG-INTERACTIES ].
Effect van antacida op de farmacokinetiek van capecitabine
Wanneer Maalox® (20 ml), een aluminiumhydroxide- en magnesiumhydroxide-bevattend antacidum, onmiddellijk na XELODA (1250 mg/m2, n=12 kankerpatiënten) werd toegediend, namen de AUC en Cmax toe met respectievelijk 16% en 35%, voor capecitabine en met respectievelijk 18% en 22% voor 5'-DFCR. Er werd geen effect waargenomen op de andere drie belangrijke metabolieten (5'-DFUR, 5-FU, FBAL) van XELODA.
Effect van allopurinol op capecitabine
Uit gepubliceerde literatuur blijkt dat gelijktijdig gebruik met allopurinol de omzetting van capecitabine in de actieve metabolieten, FdUMP en FUTP, kan verminderen; de klinische significantie was echter niet volledig gekarakteriseerd.
Effect van capecitabine op de farmacokinetiek van docetaxel en vice versa
Een fase 1-onderzoek evalueerde het effect van XELODA op de farmacokinetiek van docetaxel (Taxotere®) en het effect van docetaxel op de farmacokinetiek van XELODA werd uitgevoerd bij 26 patiënten met solide tumoren. XELODA bleek geen effect te hebben op de farmacokinetiek van docetaxel (Cmax en AUC) en docetaxel heeft geen effect op de farmacokinetiek van capecitabine en de 5-FU-precursor 5'-DFUR.
Klinische studies
Adjuvante darmkanker
Een gerandomiseerde, gecontroleerde klinische fase 3-studie in meerdere centra bij patiënten met Dukes' C colonkanker (X-ACT) leverde gegevens op over het gebruik van XELODA voor de adjuvante behandeling van patiënten met colonkanker. Het primaire doel van het onderzoek was het vergelijken van ziektevrije overleving (DFS) bij patiënten die XELODA kregen met patiënten die alleen IV 5-FU/LV kregen. In dit onderzoek werden 1987 patiënten gerandomiseerd naar behandeling met XELODA 1250 mg/m2 tweemaal daags oraal gedurende 2 weken gevolgd door een rustperiode van 1 week, gegeven als cycli van 3 weken voor in totaal 8 cycli (24 weken) of IV bolus 5-FU 425 mg/m2 en 20 mg/m2 IV leucovorine op dag 1 tot 5, gegeven als cycli van 4 weken voor in totaal 6 cycli (24 weken). Patiënten in de studie moesten tussen 18 en 75 jaar oud zijn met histologisch bevestigde Dukes' stadium C colonkanker met ten minste één positieve lymfeklier en moesten (binnen 8 weken voorafgaand aan randomisatie) volledige resectie van de primaire tumor hebben ondergaan zonder macroscopisch of microscopisch bewijs van resterende tumor. Patiënten moesten ook geen eerdere cytotoxische chemotherapie of immunotherapie hebben ondergaan (behalve steroïden), en een ECOG-prestatiestatus van 0 of 1 (KPS ≥ 70%), ANC ≥ 1,5x109/L, bloedplaatjes ≥ 100x109/L, serumcreatinine ≤ 1,5 ULN, totaal bilirubine ≤ 1,5 ULN, AST/ALT ≤ 2,5 ULN en CEA binnen de normale limieten op het moment van randomisatie.
De demografische gegevens bij baseline voor XELODA- en 5-FU/LV-patiënten worden weergegeven in Tabel 10. De kenmerken bij baseline waren goed uitgebalanceerd tussen de armen.
Alle patiënten met een normale nierfunctie of een lichte nierfunctiestoornis begonnen met de behandeling met de volledige startdosering van 1250 mg/m2 oraal tweemaal daags. De startdosis was verlaagd bij patiënten met een matige nierfunctiestoornis (berekende creatinineklaring 30 tot 50 ml/min) bij aanvang (zie DOSERING EN ADMINISTRATIE ]. Vervolgens werden voor alle patiënten de doses indien nodig aangepast op basis van de toxiciteit. Het dosisbeheer voor XELODA 500 mg omvatte dosisverlagingen, cyclusvertragingen en onderbrekingen van de behandeling (zie tabel 11).
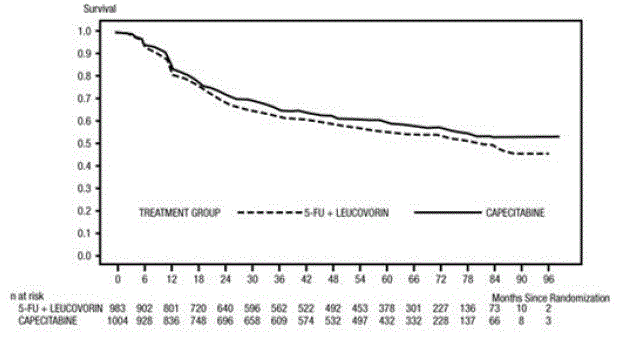
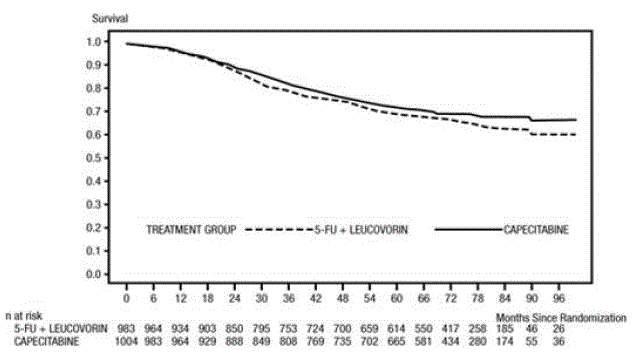
De mediane follow-up ten tijde van de analyse was 83 maanden (6,9 jaar). De hazard ratio voor DFS voor XELODA 500 mg vergeleken met 5-FU/LV was 0,88 (95% BI 0,77 – 1,01) (zie en Figuur 1 ). Omdat de bovenste 2-zijdige 95%-betrouwbaarheidslimiet van de hazard ratio lager was dan 1,20, was XELODA niet-inferieur aan 5-FU/LV. De keuze van de non-inferioriteitsmarge van 1,20 komt overeen met het behoud van ongeveer 75% van het 5-FU/LV-effect op DFS. De hazard ratio voor XELODA vergeleken met 5-FU/LV met betrekking tot de totale overleving was 0,86 (95% BI 0,74 – 1,01). De totale 5-jaarsoverleving was 71,4% voor XELODA en 68,4% voor 5-FU/LV (zie figuur 2).
Figuur 1 Kaplan-Meier-schattingen van ziektevrije overleving (alle gerandomiseerde populaties)a
Figuur 2 Kaplan-Meier-schattingen van totale overleving (alle gerandomiseerde populaties)
Gemetastaseerde colorectale kanker
Algemeen
De aanbevolen dosis XELODA 500 mg werd bepaald in een open-label, gerandomiseerde klinische studie, waarin de werkzaamheid en veiligheid van continue therapie met capecitabine (1331 mg/m2/dag in twee verdeelde doses, n=39), intermitterende therapie met capecitabine ( 2510 mg/m2/dag in twee verdeelde doses, n=34), en intermitterende therapie met capecitabine in combinatie met oraal leucovorine (LV) (capecitabine 1657 mg/m2/dag in twee verdeelde doses, n=35; leucovorine 60 mg/ dag) bij patiënten met gevorderd en/of gemetastaseerd colorectaal carcinoom in de eerstelijns gemetastaseerde setting. Er was geen duidelijk voordeel in respons op het toevoegen van leucovorine aan XELODA; de toxiciteit was echter verhoogd. XELODA, 1250 mg/m2 tweemaal daags gedurende 14 dagen gevolgd door een rustperiode van 1 week, werd geselecteerd voor verdere klinische ontwikkeling op basis van het algemene veiligheids- en werkzaamheidsprofiel van de drie onderzochte schema's.
Monotherapie
Gegevens uit twee open-label, multicenter, gerandomiseerde, gecontroleerde klinische onderzoeken met 1207 patiënten ondersteunen het gebruik van XELODA bij de eerstelijnsbehandeling van patiënten met gemetastaseerd colorectaal carcinoom. De twee klinische onderzoeken waren identiek qua opzet en werden uitgevoerd in 120 centra in verschillende landen. Studie 1 werd uitgevoerd in de VS, Canada, Mexico en Brazilië; Studie 2 werd uitgevoerd in Europa, Israël, Australië, Nieuw-Zeeland en Taiwan. In beide onderzoeken werden in totaal 603 patiënten gerandomiseerd voor behandeling met XELODA 500 mg in een dosis van 1250 mg/m2 tweemaal daags gedurende 2 weken, gevolgd door een rustperiode van 1 week en gegeven in cycli van 3 weken; 604 patiënten werden gerandomiseerd voor behandeling met 5-FU en leucovorine (20 mg/m2 leucovorine IV gevolgd door 425 mg/m2 IV bolus 5-FU, op dag 1 tot 5, elke 28 dagen).
In beide onderzoeken werden de totale overleving, de tijd tot progressie en het responspercentage (volledige plus gedeeltelijke responsen) beoordeeld. Reacties werden gedefinieerd door de criteria van de Wereldgezondheidsorganisatie en voorgelegd aan een geblindeerde onafhankelijke beoordelingscommissie (IRC). Verschillen in beoordelingen tussen de onderzoeker en IRC werden verzoend door de sponsor, geblindeerd voor de behandelingsarm, volgens een gespecificeerd algoritme. Overleving werd beoordeeld op basis van een non-inferioriteitsanalyse.
De demografische gegevens bij baseline voor XELODA- en 5-FU/LV-patiënten worden weergegeven in Tabel 13.
De werkzaamheidseindpunten voor de twee fase 3-onderzoeken worden weergegeven in en
Figuur 3 Kaplan-Meier-curve voor algehele overleving van gepoolde gegevens (onderzoeken 1 en 2)
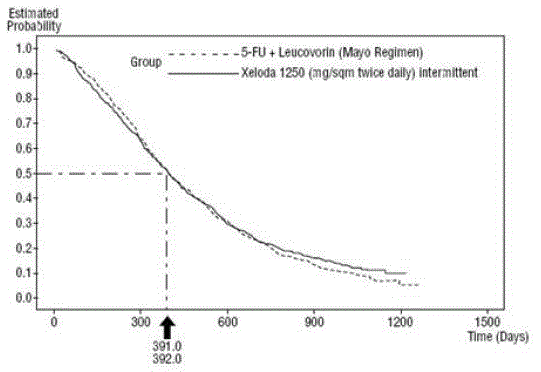
XELODA was superieur aan 5-FU/LV wat betreft het objectieve responspercentage in onderzoek 1 en onderzoek 2. De overeenkomst tussen XELODA 500 mg en 5-FU/LV in deze onderzoeken werd beoordeeld door het potentiële verschil tussen de twee behandelingen te onderzoeken. Om er zeker van te zijn dat XELODA 500 mg een klinisch betekenisvol overlevingseffect heeft, werden statistische analyses uitgevoerd om het percentage van het overlevingseffect van 5-FU/LV te bepalen dat door XELODA werd behouden. De schatting van het overlevingseffect van 5-FU/LV is afgeleid van een meta-analyse van tien gerandomiseerde onderzoeken uit de gepubliceerde literatuur waarin 5-FU werd vergeleken met regimes van 5-FU/LV die vergelijkbaar waren met de controle-armen die in deze onderzoeken werden gebruikt. 1 en 2. De methode voor het vergelijken van de behandelingen was om in het slechtste geval (95% betrouwbaarheidsbovengrens) het verschil tussen 5-FU/LV en XELODA 500 mg te onderzoeken en aan te tonen dat verlies van meer dan 50% van de 5- FU/LV overlevingseffect werd uitgesloten. Er werd aangetoond dat het percentage van het overlevingseffect van 5-FU/LV dat gehandhaafd bleef ten minste 61% was voor onderzoek 2 en 10% voor onderzoek 1. Het gepoolde resultaat komt overeen met een behoud van ten minste 50% van het effect van 5 -FU/LV. Opgemerkt moet worden dat deze waarden voor het behouden effect zijn gebaseerd op de bovengrens van het verschil tussen 5-FU/LV en XELODA 500 mg. Deze resultaten sluiten de mogelijkheid van echte equivalentie van XELODA 500 mg met 5-FU/LV niet uit (zie en .). figuur 3 ).
Borstkanker
XELODA is geëvalueerd in klinische onderzoeken in combinatie met docetaxel (Taxotere®) en als monotherapie.
In combinatie met Docetaxel
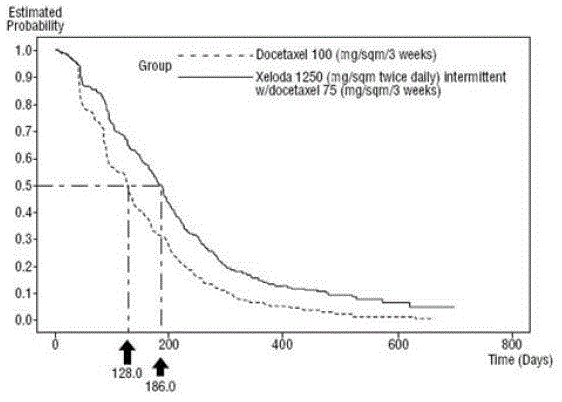
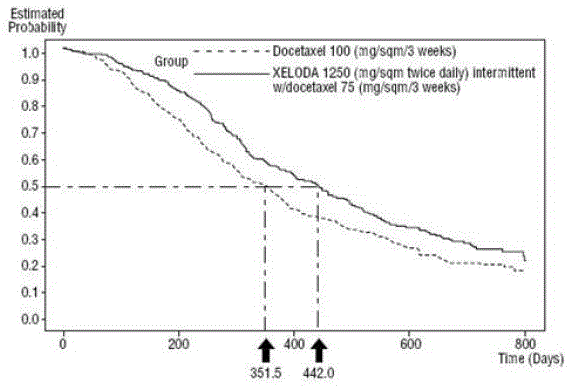
De dosis XELODA die werd gebruikt in het klinische fase 3-onderzoek in combinatie met docetaxel was gebaseerd op de resultaten van een fase 1-onderzoek, waarbij een reeks doses docetaxel toegediend in cycli van 3 weken in combinatie met een intermitterend regime van XELODA (14 dagen behandeling, gevolgd door een rustperiode van 7 dagen) werden geëvalueerd. Het combinatiedoseringsschema werd gekozen op basis van het verdraagbaarheidsprofiel van de 75 mg/m2 toegediend in cycli van 3 weken van docetaxel in combinatie met 1250 mg/m2 tweemaal daags gedurende 14 dagen XELODA 500 mg toegediend in cycli van 3 weken. De goedgekeurde dosis van 100 mg/m2 docetaxel toegediend in cycli van 3 weken was de controle-arm van het fase 3-onderzoek.
XELODA in combinatie met docetaxel werd beoordeeld in een open-label, multicenter, gerandomiseerde studie in 75 centra in Europa, Noord-Amerika, Zuid-Amerika, Azië en Australië. Een totaal van 511 patiënten met gemetastaseerde borstkanker die resistent was tegen, of terugkeerde tijdens of na een antracycline-bevattende therapie, of recidiverend tijdens of recidiverend binnen 2 jaar na voltooiing van een antracycline-bevattende adjuvante therapie, werden geïncludeerd. Tweehonderdvijfenvijftig (255) patiënten werden gerandomiseerd naar XELODA 1250 mg/m2 tweemaal daags gedurende 14 dagen gevolgd door 1 week zonder behandeling en docetaxel 75 mg/m2 als een 1-uurs intraveneuze infusie toegediend in cycli van 3 weken. In de monotherapiearm kregen 256 patiënten docetaxel 100 mg/m2 als een 1-uurs intraveneuze infusie toegediend in cycli van 3 weken. Demografische gegevens van patiënten worden gegeven in tabel 16.
XELODA in combinatie met docetaxel resulteerde in een statistisch significante verbetering in tijd tot ziekteprogressie, algehele overleving en objectieve respons in vergelijking met monotherapie met docetaxel zoals getoond in en Figuur 5 .
Figuur 4 Kaplan-Meier-schattingen voor tijd tot ziekteprogressie XELODA en Docetaxel vs Docetaxel
Figuur 5 Kaplan-Meier schattingen van overleving XELODA en Docetaxel vs Docetaxel
Monotherapie
De antitumoractiviteit van XELODA als monotherapie werd geëvalueerd in een open-label eenarmige studie die werd uitgevoerd in 24 centra in de VS en Canada. In totaal werden 162 patiënten met stadium IV borstkanker geïncludeerd. Het primaire eindpunt was het tumorresponspercentage bij patiënten met meetbare ziekte, met een respons gedefinieerd als een ≥50% afname van de som van de producten van de loodrechte diameters van bidimensionaal meetbare ziekte gedurende ten minste 1 maand. XELODA werd toegediend in een dosis van 1255 mg/m2 tweemaal daags gedurende 2 weken, gevolgd door een rustperiode van 1 week en gegeven als cycli van 3 weken. De demografische gegevens en klinische kenmerken bij baseline voor alle patiënten (n=162) en patiënten met meetbare ziekte (n=135) worden weergegeven in . Resistentie werd gedefinieerd als progressieve ziekte tijdens de behandeling, met of zonder een initiële respons, of terugval binnen 6 maanden na voltooiing van de behandeling met een antracycline-bevattend adjuvans chemotherapieregime.
Antitumorresponsen voor patiënten met een ziekte die resistent is tegen zowel paclitaxel als een antracycline worden getoond in:
Voor de subgroep van 43 patiënten die dubbel resistent waren, was de mediane tijd tot progressie 102 dagen en de mediane overleving was 255 dagen. Het objectieve responspercentage in deze populatie werd ondersteund door een responspercentage van 18,5% (1 CR, 24 PR's) in de totale populatie van 135 patiënten met meetbare ziekte, die minder resistent waren tegen chemotherapie (zie ). De mediane tijd tot progressie was 90 dagen en de mediane overleving was 306 dagen.
PATIËNT INFORMATIE
XELODA® (zeh-LOE-duh) (capecitabine) tabletten
Wat is de belangrijkste informatie die ik moet weten over XELODA 500 mg?
XELODA 500 mg kan ernstige bijwerkingen veroorzaken, waaronder:
- XELODA kan een wisselwerking hebben met bloedverdunners, zoals warfarine (COUMADIN®). Het gebruik van XELODA 500 mg met deze geneesmiddelen kan veranderingen veroorzaken in hoe snel uw bloed stolt en kan bloedingen veroorzaken die tot de dood kunnen leiden. Dit kan gebeuren zodra u bent begonnen met het innemen van XELODA 500 mg, of later tijdens de behandeling, en mogelijk zelfs binnen 1 maand nadat u bent gestopt met het innemen van XELODA. Uw risico kan hoger zijn omdat u kanker heeft en als u ouder bent dan 60 jaar.
- Vertel uw zorgverlener voordat u XELODA inneemt als u warfarine (COUMADIN) of een ander bloedverdunnermiddel gebruikt.
- Als u warfarine (COUMADIN) of een andere bloedverdunner die lijkt op warfarine (COUMADIN) gebruikt tijdens de behandeling met XELODA, moet uw zorgverlener vaak bloedonderzoek doen om te controleren hoe snel uw bloed stolt tijdens en nadat u met de behandeling met XELODA bent gestopt. Uw zorgverlener kan uw dosis van het bloedverdunnende geneesmiddel indien nodig wijzigen.
Zien “Wat zijn de mogelijke bijwerkingen van XELODA 500 mg?” voor meer informatie over bijwerkingen.
Wat is XELODA?
XELODA is een receptgeneesmiddel dat wordt gebruikt voor de behandeling van mensen met:
- kanker van de dikke darm die is uitgezaaid naar de lymfeklieren in het gebied dicht bij de dikke darm (stadium Dukes' C), nadat ze een operatie hebben ondergaan.
- kanker van de dikke darm of het rectum (colorectaal) die is uitgezaaid naar andere delen van het lichaam (gemetastaseerd), als uw eerste behandeling van uw kanker in dit stadium.
- borstkanker die is uitgezaaid naar andere delen van het lichaam (gemetastaseerd) samen met een ander geneesmiddel, docetaxel genaamd, nadat behandeling met bepaalde andere geneesmiddelen tegen kanker niet heeft gewerkt.
- borstkanker die zich verspreidt naar andere delen van het lichaam en niet is verbeterd na behandeling met paclitaxel en bepaalde andere geneesmiddelen tegen kanker, of die geen behandeling meer kunnen krijgen met bepaalde geneesmiddelen tegen kanker.
Het is niet bekend of XELODA 500 mg veilig en effectief is bij kinderen.
Gebruik XELODA niet als u:
- ernstige nierproblemen hebben
- zijn allergisch voor capecitabine, 5-fluorouracil of voor één van de bestanddelen van XELODA. Zie het einde van deze bijsluiter voor een volledige lijst van ingrediënten in XELODA.
Neem contact op met uw zorgverlener voordat u XELODA gebruikt als u niet zeker weet of u een van de hierboven genoemde aandoeningen heeft.
Vertel uw zorgverlener voordat u XELODA gebruikt over al uw medische aandoeningen, ook als u:
Zien “Wat is de belangrijkste informatie die ik over XELODA moet weten?”
- hartproblemen hebben gehad
- nier- of leverproblemen heeft
- is verteld dat u het enzym DPD (dihydropyrimidinedehydrogenase) mist.
- zwanger bent of van plan bent zwanger te worden. XELODA 500 mg kan uw ongeboren baby schaden. Uw zorgverlener moet een zwangerschapstest doen voordat u begint met de behandeling met XELODA. Vertel het uw zorgverlener meteen als u zwanger wordt of denkt dat u zwanger zou kunnen zijn tijdens de behandeling met XELODA.
- vrouwen die zwanger kunnen worden, moeten effectieve anticonceptie gebruiken tijdens de behandeling en gedurende 6 maanden na de laatste dosis. Praat met uw zorgverlener over anticonceptiekeuzes die voor u geschikt kunnen zijn tijdens de behandeling met XELODA.
- mannen die vrouwelijke partners hebben die zwanger kunnen worden, moeten effectieve anticonceptie gebruiken tijdens de behandeling en gedurende 3 maanden na de laatste dosis.
- borstvoeding geeft of van plan bent borstvoeding te geven. Het is niet bekend of XELODA in uw moedermelk terechtkomt. Geef geen borstvoeding tijdens de behandeling met XELODA 500 mg en gedurende 2 weken na de laatste dosis.
Vertel uw zorgverlener over alle geneesmiddelen die u gebruikt, inclusief geneesmiddelen op recept en vrij verkrijgbare medicijnen, vitamines en kruidensupplementen. XELODA kan de werking van andere geneesmiddelen beïnvloeden en andere geneesmiddelen kunnen de werking van XELODA beïnvloeden.
Weet welke medicijnen u gebruikt. Houd er een lijst van bij om uw zorgverlener en apotheker te laten zien wanneer u een nieuw geneesmiddel krijgt.
Hoe moet ik XELODA 500 mg innemen?
- Neem XELODA precies in zoals uw zorgverlener u zegt dat u het moet innemen.
- Uw zorgverlener zal u vertellen hoeveel XELODA u moet innemen en wanneer u het moet innemen.
- Neem XELODA 2 keer per dag, 1 keer 's ochtends en 1 keer 's avonds.
- Neem XELODA 500 mg binnen 30 minuten na het beëindigen van een maaltijd in.
- Slik XELODA 500 mg tabletten heel door met water. Niet doen maal of snijd XELODA 500 mg tabletten. Vertel het uw arts als u XELODA-tabletten niet heel kunt doorslikken.
- Uw zorgverlener kan uw dosis veranderen, tijdelijk stoppen of permanent stoppen met de behandeling met XELODA 500 mg als u bijwerkingen krijgt.
- Als u te veel XELODA heeft ingenomen, neem dan onmiddellijk contact op met uw zorgverlener of ga naar de eerste hulpafdeling van het dichtstbijzijnde ziekenhuis.
Wat zijn de mogelijke bijwerkingen van XELODA?
XELODA kan ernstige bijwerkingen veroorzaken, waaronder:
Misselijkheid en braken komen vaak voor bij XELODA. Als u uw eetlust verliest, zich zwak voelt en misselijkheid, braken of diarree heeft, kunt u snel uitgedroogd raken.
Stop met het gebruik van XELODA en bel onmiddellijk uw zorgverlener als u:
Als uw aantal witte bloedcellen erg laag is, loopt u een verhoogd risico op infectie. Bel onmiddellijk uw zorgverlener als u koorts krijgt van 100, 5 ° F of hoger of andere tekenen en symptomen van infectie heeft.
- Zien “Wat is de belangrijkste informatie die ik over XELODA moet weten?”
- Diarree. Diarree komt vaak voor bij XELODA en kan soms ernstig zijn. Stop met het innemen van XELODA en bel onmiddellijk uw zorgverlener als het aantal stoelgangen dat u per dag heeft met 4 of meer stoelgangen toeneemt dan gebruikelijk voor u of 's nachts stoelgang. Vraag uw zorgverlener welke medicijnen u kunt nemen om uw diarree te behandelen. Als u ernstige bloederige diarree heeft met ernstige buikpijn en koorts, stop dan met het gebruik van Xeloda 500 mg en bel uw zorgverlener of ga onmiddellijk naar de eerste hulp van het dichtstbijzijnde ziekenhuis.
- Hart problemen. XELODA kan hartproblemen veroorzaken, waaronder: hartaanval en verminderde bloedtoevoer naar het hart, pijn op de borst, onregelmatige hartslag, veranderingen in de elektrische activiteit van uw hart gezien op een elektrocardiogram (ECG), problemen met uw hartspier, hartfalen en plotselinge dood. Stop met het innemen van XELODA 500 mg en bel uw zorgverlener of ga direct naar de dichtstbijzijnde spoedeisende hulp van het ziekenhuis als u nieuwe symptomen van een hartprobleem krijgt, waaronder:
- pijn op de borst
- duizeligheid
- kortademigheid
- duizeligheid
- Verlies van te veel lichaamsvloeistof (uitdroging) en nierfalen. Uitdroging kan optreden met XELODA 500 mg en kan plotseling nierfalen veroorzaken dat tot de dood kan leiden. U loopt een groter risico als u nierproblemen heeft voordat u XELODA 500 mg inneemt en u ook andere geneesmiddelen gebruikt die nierproblemen kunnen veroorzaken.
- 2 of meer keer per dag overgeven.
- slechts af en toe wat kunnen eten of drinken, of helemaal niet door misselijkheid.
- diarree hebben. Zie "diarree" hierboven.
- Ernstige huid- en mondreacties.
- XELODA 500 mg kan ernstige huidreacties veroorzaken die tot de dood kunnen leiden. Vertel het uw zorgverlener meteen als u huiduitslag, blaren en vervelling van uw huid krijgt. Uw zorgverlener kan u vertellen om te stoppen met het gebruik van XELODA als u een ernstige huidreactie krijgt. Neem XELODA 500 mg niet meer in als dit gebeurt.
- XELODA 500 mg kan ook het "hand-en-voet"-syndroom veroorzaken. Het hand- en voetsyndroom komt vaak voor bij XELODA 500 mg en kan ervoor zorgen dat u gevoelloosheid en veranderingen in het gevoel in uw handen en voeten krijgt, of roodheid, pijn, zwelling van uw handen en voeten. Stop met het gebruik van XELODA en bel onmiddellijk uw zorgverlener als u een van deze symptomen heeft en u niet in staat bent uw gebruikelijke activiteiten uit te voeren.
- Hand-en-voetsyndroom kan leiden tot verlies van vingerafdrukken, wat van invloed kan zijn op uw identificatie.
- kunt zweren in uw mond of op uw tong krijgen als u XELODA gebruikt. Stop met het gebruik van XELODA 500 mg en bel uw zorgverlener als u pijnlijke roodheid, zwelling of zweren in uw mond of tong krijgt, of als u problemen heeft met eten.
- Verhoogd bilirubinegehalte in uw bloed en leverproblemen. Verhoogd bilirubine in uw bloed komt vaak voor bij XELODA 500 mg en kan soms ook ernstig zijn. Uw zorgverlener zal u tijdens de behandeling met XELODA op deze problemen controleren.
- Verlaagd aantal witte bloedcellen, bloedplaatjes en rode bloedcellen. Uw zorgverlener zal tijdens de behandeling met XELODA bloedonderzoek doen om uw bloedceltellingen te controleren.
Mensen van 80 jaar of ouder kunnen een grotere kans hebben om ernstige of ernstige bijwerkingen te krijgen met XELODA.
De meest voorkomende bijwerkingen van XELODA 500 mg zijn:
- diarree
- maagstreek (buikpijn)
- hand- en voetsyndroom
- zwakte en vermoeidheid
- misselijkheid
- overgegeven
- verhoogde hoeveelheden afbraakproducten van rode bloedcellen (bilirubine in uw bloed)
Ernstige allergische reacties kunnen optreden met XELODA. Vertel het uw zorgverlener als u ooit een allergische reactie heeft gehad op capecitabine of 5-fluorouracil. Zien “Neem XELODA niet in als u:”. Stop met het innemen van XELODA 500 mg en bel onmiddellijk uw zorgverlener of ga naar de eerste hulp als u een van de volgende symptomen van een ernstige allergische reactie heeft:
- rode jeukende striemen op uw huid (netelroos)
- roodheid van de huid
- zwelling van uw gezicht, lippen, tong of keel
- uitslag
- jeuk
- moeite met slikken of ademen
XELODA 500 mg kan vruchtbaarheidsproblemen veroorzaken bij vrouwen en mannen. Dit kan van invloed zijn op het vermogen om een kind te krijgen. Praat met uw zorgverlener als u zich zorgen maakt over de vruchtbaarheid.
Dit zijn niet alle mogelijke bijwerkingen van XELODA.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
Hoe moet ik XELODA bewaren?
- Bewaar XELODA 500 mg bij kamertemperatuur tussen 20 °C en 25 °C (68 °F tot 77 °F).
- Bewaar XELODA 500 mg in een goed gesloten container.
- Vraag uw zorgverlener of apotheker hoe u ongebruikte XELODA veilig weggooit.
Houd XELODA en alle geneesmiddelen buiten het bereik van kinderen.
Algemene informatie over het veilige en effectieve gebruik van XELODA.
Geneesmiddelen worden soms voorgeschreven voor andere doeleinden dan vermeld in een patiëntenbijsluiter. Gebruik XELODA niet voor een aandoening waarvoor het niet is voorgeschreven. Geef XELODA 500 mg niet aan andere mensen, ook niet als zij dezelfde symptomen hebben als u. Het kan hen schaden. U kunt uw apotheker of zorgverlener om informatie vragen over XELODA 500 mg die is geschreven voor gezondheidswerkers.
Wat zijn de ingrediënten in XELODA 500 mg?
Actief ingrediënt: capecitabine
Inactieve ingredienten: watervrije lactose, croscarmellosenatrium, hydroxypropylmethylcellulose, microkristallijne cellulose, magnesiumstearaat en gezuiverd water. De perzikkleurige of licht perzikkleurige filmomhulling bevat hydroxypropylmethylcellulose, talk, titaandioxide en synthetische gele en rode ijzeroxiden.
Ga voor meer informatie naar http://www.gene.com/patients/medicines/xeloda 500 mg of bel 1-877-436-3683.
Deze patiëntinformatie is goedgekeurd door de Amerikaanse Food and Drug Administration.