Tricor 160mg, 200mg Fenofibrate Gebruik, bijwerkingen en dosering. Prijs in online apotheek. Generieke medicijnen zonder recept.
Wat is Tricor en hoe wordt het gebruikt?
Tricor is een receptgeneesmiddel dat wordt gebruikt om de symptomen van cholesterol en triglyceriden (vetzuren) in het bloed te verminderen. Tricor kan alleen of met andere medicijnen worden gebruikt.
- Tricor behoort tot een klasse geneesmiddelen die Fibric Acid Agents worden genoemd.
- Het is niet bekend of Tricor 200 mg veilig en effectief is bij kinderen
Wat zijn de mogelijke bijwerkingen van Tricor?
Tricor 200 mg kan ernstige bijwerkingen veroorzaken, waaronder:
- scherpe buikpijn die zich uitbreidt naar uw rug of schouderblad,
- verlies van eetlust,
- maagpijn na een maaltijd,
- geel worden van de huid of ogen (geelzucht),
- koorts,
- rillingen,
- zwakheid,
- keelpijn,
- zweertjes in de mond,
- ongewone blauwe plekken of bloedingen,
- pijn op de borst,
- plotselinge hoest,
- piepende ademhaling,
- snel ademhalen,
- bloed ophoesten, en
- zwelling, warmte of roodheid in een arm of been
Roep meteen medische hulp in als u een van de bovenstaande symptomen heeft.
De meest voorkomende bijwerkingen van Tricor zijn:
- loopneus,
- niezen, en
- abnormale laboratoriumtests
Vertel het uw arts als u een bijwerking heeft die u hindert of die niet weggaat.
Dit zijn niet alle mogelijke bijwerkingen van Tricor. Vraag uw arts of apotheker om meer informatie.
Bel uw arts voor medisch advies over bijwerkingen. U kunt bijwerkingen melden aan de FDA op 1-800-FDA-1088.
OMSCHRIJVING
TRICOR (fenofibraattabletten), is een lipidenregulerend middel dat verkrijgbaar is als tabletten voor orale toediening. Elke tablet bevat 54 mg of 160 mg fenofibraat. De chemische naam voor fenofibraat is 2-[4-(4-chloorbenzoyl)fenoxy]-2-methylpropaanzuur, 1-methylethylester met de volgende structuurformule:
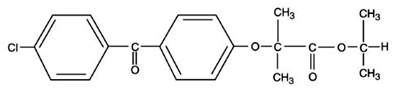
De empirische formule is C20H21O4Cl en het molecuulgewicht is 360,83; fenofibraat is onoplosbaar in water. Het smeltpunt is 79-82°C. Fenofibraat is een witte vaste stof die onder normale omstandigheden stabiel is.
inactieve ingredienten
Elke tablet bevat colloïdaal siliciumdioxide, crospovidon, lactosemonohydraat, lecithine, microkristallijne cellulose, polyvinylalcohol, povidon, natriumlaurylsulfaat, natriumstearylfumaraat, talk, titaniumdioxide en xanthaangom. Daarnaast bevatten individuele tabletten van 54 mg D&C Yellow No. 10, FD&C Yellow No. 6, FD&C Blue No. 2.
INDICATIES
Primaire hypercholesterolemie of gemengde dyslipidemie
TRICOR is geïndiceerd als aanvullende therapie bij een dieet om verhoogde lipoproteïne-cholesterol met lage dichtheid (LDL-C), totaal cholesterol (Total-C), triglyceriden en apolipoproteïne B (Apo B) te verlagen en om high-density lipoproteïne-cholesterol (HDL- C) bij volwassen patiënten met primaire hypercholesterolemie of gemengde dyslipidemie.
Ernstige hypertriglyceridemie
TRICOR 200 mg is ook geïndiceerd als aanvullende therapie bij een dieet voor de behandeling van volwassen patiënten met ernstige hypertriglyceridemie. Verbetering van de glykemische controle bij diabetespatiënten die nuchtere chylomicronemie vertonen, zal gewoonlijk de noodzaak van farmacologische interventie wegnemen.
Duidelijk verhoogde niveaus van serumtriglyceriden (bijv. > 2.000 mg/dL) kunnen het risico op het ontwikkelen van pancreatitis verhogen. Het effect van behandeling met fenofibraat op het verminderen van dit risico is niet voldoende onderzocht.
Belangrijke gebruiksbeperkingen
Fenofibraat in een dosis gelijk aan 145 mg TRICOR bleek de morbiditeit en mortaliteit van coronaire hartziekten niet te verminderen in een groot, gerandomiseerd gecontroleerd onderzoek bij patiënten met type 2 diabetes mellitus [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
DOSERING EN ADMINISTRATIE
Algemene Overwegingen
Patiënten moeten op een geschikt lipidenverlagend dieet worden gezet voordat ze TRICOR 160 mg krijgen, en moeten dit dieet voortzetten tijdens de behandeling met TRICOR. TRICOR-tabletten kunnen worden gegeven zonder rekening te houden met maaltijden.
De initiële behandeling voor dyslipidemie is een dieettherapie die specifiek is voor het type lipoproteïne-afwijking. Overmatig lichaamsgewicht en overmatige alcoholinname kunnen belangrijke factoren zijn bij hypertriglyceridemie en moeten vóór elke medicamenteuze behandeling worden aangepakt. Lichaamsbeweging kan een belangrijke aanvullende maatregel zijn. Ziekten die bijdragen aan hyperlipidemie, zoals hypothyreoïdie of diabetes mellitus, moeten worden opgespoord en adequaat worden behandeld. Oestrogeentherapie, thiazidediuretica en bètablokkers gaan soms gepaard met enorme stijgingen van plasmatriglyceriden, vooral bij personen met familiaire hypertriglyceridemie. In dergelijke gevallen kan stopzetting van het specifieke etiologische middel de noodzaak van specifieke medicamenteuze behandeling van hypertriglyceridemie overbodig maken.
Lipidespiegels moeten periodiek worden gecontroleerd en er moet worden overwogen de dosering van TRICOR te verlagen als de lipidespiegels aanzienlijk onder het beoogde bereik dalen.
Bij patiënten die na twee maanden behandeling geen adequate respons hebben, moet de therapie worden stopgezet met de maximaal aanbevolen dosis van 145 mg eenmaal daags.
Primaire hypercholesterolemie of gemengde dyslipidemie
De aanvangsdosis van TRICOR 200 mg is eenmaal daags 145 mg.
Ernstige hypertriglyceridemie
De aanvangsdosis is 48 tot 145 mg per dag. De dosering moet individueel worden aangepast aan de respons van de patiënt en moet indien nodig worden aangepast na herhaalde lipidebepalingen met tussenpozen van 4 tot 8 weken. De maximale dosis is 145 mg eenmaal daags.
Verminderde nierfunctie
Behandeling met TRICOR 200 mg dient te worden gestart met een dosis van 48 mg per dag bij patiënten met een lichte tot matige nierfunctiestoornis, en mag pas worden verhoogd na evaluatie van de effecten op de nierfunctie en de lipidenspiegels bij deze dosis. Het gebruik van TRICOR moet worden vermeden bij patiënten met een ernstige nierfunctiestoornis [zie: Gebruik bij specifieke populaties en KLINISCHE FARMACOLOGIE ].
Geriatrische patiënten
Dosiskeuze voor ouderen moet worden gemaakt op basis van de nierfunctie [zie Gebruik bij specifieke populaties ].
HOE GELEVERD
Doseringsvormen en sterke punten
- Gele tabletten van 48 mg, bedrukt met de code-identificatieletters “FI”.
- Gele tabletten van 48 mg, bedrukt met het “a”-logo en de code-identificatieletters “FI”.
- Witte tabletten van 145 mg, bedrukt met de code-identificatieletters “FO”.
- Witte tabletten van 145 mg, bedrukt met het “a”-logo en de code-identificatieletters “FO”.
Opslag en behandeling
TRICOR® (fenofibraattabletten) is verkrijgbaar in twee sterktes:
48 mg
Gele tabletten, bedrukt met de code-identificatieletters "FI", verkrijgbaar in flessen van 90 ( NDC 0074-3173-90).
Gele tabletten, bedrukt met het "a"-logo en code-identificatieletters "FI", verkrijgbaar in flessen van 90 ( NDC 0074-6122-90).
145 mg
Witte tabletten, bedrukt met de code-identificatieletters "FO", verkrijgbaar in flessen van 90 ( NDC 0074-3189-90).
Witte tabletten, bedrukt met het "a"-logo en code-identificatieletters "FO", verkrijgbaar in flessen van 90 ( NDC 0074-6123-90).
Opslag
Bewaren bij 25°C (77°F); excursies toegestaan tot 15-30 ° C (59-86 ° F).
[Zie USP-gecontroleerde kamertemperatuur]. Buiten bereik van kinderen bewaren. Beschermen tegen vocht.
Gefabriceerd voor AbbVie Inc., North Chicago, IL 60064, VS door Fournier Laboratories Ireland Limited, Anngrove, Carrigtwohill Co. Cork, Ierland. Herzien : maart 2021
BIJWERKINGEN
De volgende ernstige bijwerkingen worden hieronder en elders in de etikettering beschreven:
- Mortaliteit en coronaire hartziekte morbiditeit [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
- levertoxiciteit [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
- Pancreatitis [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
- Overgevoeligheidsreacties [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
- Venotrombo-embolische ziekte [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]
Ervaring met klinische proeven
Omdat klinische onderzoeken onder sterk uiteenlopende omstandigheden worden uitgevoerd, kunnen de bijwerkingen die in de klinische onderzoeken van een geneesmiddel zijn waargenomen niet direct worden vergeleken met de percentages in de klinische onderzoeken van een ander geneesmiddel en komen mogelijk niet overeen met de in de praktijk waargenomen percentages.
Bijwerkingen die werden gemeld door 2% of meer van de patiënten die werden behandeld met fenofibraat (en meer dan placebo) tijdens de dubbelblinde, placebogecontroleerde onderzoeken, ongeacht de causaliteit, staan vermeld in tabel 1 hieronder. Bijwerkingen leidden tot stopzetting van de behandeling bij 5,0% van de met fenofibraat behandelde patiënten en bij 3,0% die met placebo werden behandeld. Verhogingen van de leverfunctietesten waren de meest voorkomende gebeurtenissen, waardoor de behandeling met fenofibraat werd stopgezet bij 1,6% van de patiënten in dubbelblinde onderzoeken.
Urticaria werd gezien bij 1,1% vs. 0% en huiduitslag bij respectievelijk 1,4% vs. 0,8% van de fenofibraat- en placebopatiënten in gecontroleerde onderzoeken.
Toename van leverenzymen
In een gepoolde analyse van 10 placebogecontroleerde onderzoeken traden verhogingen tot > 3 maal de bovengrens van normaal in ALT op bij 5,3% van de patiënten die fenofibraat gebruikten in doses gelijk aan 96 mg tot 145 mg TRICOR per dag versus 1,1% van de patiënten behandeld met placebo [zien WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ]. In een 8 weken durende studie was de incidentie van ALAT- of ASAT-verhogingen ≥ 3 maal de bovengrens van normaal 13% bij patiënten die doseringen kregen gelijk aan 96 mg tot 145 mg TRICOR per dag en was 0% bij diegenen die doseringen kregen gelijk aan 48 mg of minder TRICOR per dag of placebo.
Postmarketingervaring
De volgende bijwerkingen zijn vastgesteld tijdens het gebruik van fenofibraat na goedkeuring. Omdat deze reacties vrijwillig worden gemeld door een populatie van onbekende grootte, is het niet altijd mogelijk om een betrouwbare schatting te maken van hun frequentie of een oorzakelijk verband te leggen met blootstelling aan geneesmiddelen: myalgie, rabdomyolyse, pancreatitis, acuut nierfalen, spierspasmen, hepatitis, cirrose, verhoogde totaal bilirubine, anemie, artralgie, afname van hemoglobine, afname van hematocriet, afname van witte bloedcellen, asthenie, ernstig verlaagde HDL-cholesterolspiegels en interstitiële longziekte. Fotosensitiviteitsreacties zijn dagen tot maanden na de start opgetreden; in sommige van deze gevallen rapporteerden patiënten een eerdere fotosensitiviteitsreactie op ketoprofen.
DRUG-INTERACTIES
Coumarine anticoagulantia
Versterking van de anticoagulerende effecten van het cumarine-type is waargenomen bij verlenging van de PT/INR.
Voorzichtigheid is geboden wanneer cumarine-anticoagulantia worden gegeven in combinatie met TRICOR. De dosering van de anticoagulantia moet worden verlaagd om de PT/INR op het gewenste niveau te houden om bloedingscomplicaties te voorkomen. Frequente PT/INR-bepalingen zijn aan te raden totdat definitief is vastgesteld dat de PT/INR gestabiliseerd is [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
Immunosuppressiva
Immunosuppressiva zoals ciclosporine en tacrolimus kunnen nefrotoxiciteit veroorzaken met een afname van de creatinineklaring en een stijging van het serumcreatinine, en omdat uitscheiding via de nieren de primaire eliminatieroute is van fibraten, waaronder TRICOR, bestaat het risico dat een interactie leidt tot verslechtering van de nierfunctie. De voordelen en risico's van het gebruik van TRICOR (fenofibraattabletten) met immunosuppressiva en andere potentieel nefrotoxische middelen moeten zorgvuldig worden overwogen, en de laagste effectieve dosis die wordt gebruikt en de nierfunctie moeten worden gecontroleerd.
Galzuurbindende harsen
Aangezien galzuurbindende harsen andere gelijktijdig toegediende geneesmiddelen kunnen binden, moeten patiënten TRICOR 200 mg ten minste 1 uur vóór of 4 tot 6 uur na een galzuurbindend hars innemen om te voorkomen dat de absorptie ervan wordt belemmerd.
Colchicine
Gevallen van myopathie, waaronder rabdomyolyse, zijn gemeld met fenofibraat gelijktijdig toegediend met colchicine, en voorzichtigheid is geboden bij het voorschrijven van fenofibraat met colchicine.
WAARSCHUWINGEN
Inbegrepen als onderdeel van de PREVENTIEVE MAATREGELEN sectie.
PREVENTIEVE MAATREGELEN
Mortaliteit en coronaire hartziekte Morbiditeit
Het effect van TRICOR 160 mg op de morbiditeit en mortaliteit van coronaire hartziekten en niet-cardiovasculaire mortaliteit is niet vastgesteld.
De Action to Control Cardiovascular Risk in Diabetes Lipid (ACCORD Lipid)-studie was een gerandomiseerde, placebogecontroleerde studie bij 5518 patiënten met type 2 diabetes mellitus die een statine als achtergrondbehandeling kregen en die werden behandeld met fenofibraat. De gemiddelde duur van de follow-up was 4,7 jaar. Fenofibraat plus statine combinatietherapie liet een niet-significante 8% relatieve risicoreductie zien in de primaire uitkomst van ernstige cardiovasculaire bijwerkingen (MACE), een samenstelling van niet-fataal myocardinfarct, niet-fatale beroerte en overlijden door cardiovasculaire ziekte (hazard ratio [ HR] 0,92, 95% BI 0,79-1,08 (p=0,32) in vergelijking met statinemonotherapie. In een geslachtssubgroepanalyse was de hazard ratio voor MACE bij mannen die combinatietherapie kregen versus statine monotherapie 0,82 (95% BI 0,69-0,99), en de hazard ratio voor MACE bij vrouwen die combinatietherapie kregen versus statine monotherapie was 1,38 (95% BI 0,98-1,94) (interactie p=0,01). De klinische betekenis van deze subgroepbevinding is onduidelijk.
De Fenofibrate Intervention and Event Lowering in Diabetes (FIELD)-studie was een 5 jaar durende, gerandomiseerde, placebogecontroleerde studie van 9795 patiënten met diabetes mellitus type 2 die werden behandeld met fenofibraat. Fenofibraat vertoonde een niet-significante 11% relatieve reductie in de primaire uitkomst van coronaire hartziekten (hazard ratio [HR] 0,89, 95% BI 0,75-1,05, p=0,16) en een significante 11% reductie in de secundaire uitkomst van totaal voorvallen van hart- en vaatziekten (HR 0,89 [0,80-0,99], p=0,04). Er was een niet-significante toename van respectievelijk 11% (HR 1,11 [0,95, 1,29], p=0,18) en 19% (HR 1,19 [0,90, 1,57], p=0,22) in de totale en coronaire hartziektesterfte met fenofibraat. in vergelijking met placebo.
Vanwege chemische, farmacologische en klinische overeenkomsten tussen TRICOR (fenofibraattabletten), clofibraat en gemfibrozil, kunnen de nadelige bevindingen in 4 grote gerandomiseerde, placebogecontroleerde klinische onderzoeken met deze andere fibraatgeneesmiddelen ook van toepassing zijn op TRICOR.
In het Coronary Drug Project, een groot onderzoek naar post-myocardinfarct van patiënten die gedurende 5 jaar met clofibraat werden behandeld, werd geen verschil in mortaliteit gezien tussen de clofibraatgroep en de placebogroep. Er was echter een verschil in het percentage cholelithiasis en cholecystitis waarvoor een operatie nodig was tussen de twee groepen (3,0% vs. 1,8%).
In een studie uitgevoerd door de Wereldgezondheidsorganisatie (WHO) werden 5000 proefpersonen zonder bekende coronaire hartziekte gedurende 5 jaar behandeld met placebo of clofibraat en nog een jaar gevolgd. Er was een statistisch significante, voor leeftijd gecorrigeerde mortaliteit door alle oorzaken in de clofibraatgroep vergeleken met de placebogroep (5,70% vs. 3,96%, p =
De Helsinki Heart Study was een grote (n=4081) studie van mannen van middelbare leeftijd zonder een voorgeschiedenis van coronaire hartziekte. De proefpersonen kregen gedurende 5 jaar placebo of gemfibrozil, met daarna een open verlenging van 3,5 jaar. De totale mortaliteit was numeriek hoger in de gemfibrozil-randomisatiegroep, maar bereikte geen statistische significantie (p = 0,19, 95% betrouwbaarheidsinterval voor relatief risico G:P = 0,91-1,64). Hoewel sterfgevallen door kanker hoger waren in de gemfibrozil-groep (p = 0,11), werden kankers (exclusief basaalcelcarcinoom) in beide onderzoeksgroepen met gelijke frequentie gediagnosticeerd. Vanwege de beperkte omvang van het onderzoek bleek het relatieve risico op overlijden door welke oorzaak dan ook niet anders te zijn dan dat in de 9 jaar durende follow-upgegevens van het onderzoek van de Wereldgezondheidsorganisatie (RR=1,29).
Een secundaire preventiecomponent van de Helsinki Heart Study omvatte mannen van middelbare leeftijd die waren uitgesloten van de primaire preventiestudie vanwege bekende of vermoede coronaire hartziekte. De proefpersonen kregen gedurende 5 jaar gemfibrozil of placebo. Hoewel de trend van hartsterfte hoger was in de gemfibrozilgroep, was dit niet statistisch significant (hazard ratio 2,2, 95% betrouwbaarheidsinterval: 0,94-5,05). Het percentage galblaasoperaties was niet statistisch significant tussen de onderzoeksgroepen, maar vertoonde wel een hogere trend in de gemfibrozilgroep (1,9% vs. 0,3%, p = 0,07).
Hepatotoxiciteit
Ernstige geneesmiddelgeïnduceerde leverbeschadiging (DILI), waaronder levertransplantatie en overlijden, is gemeld na het op de markt brengen van TRICOR. DILI is gemeld binnen de eerste paar weken van de behandeling of na enkele maanden therapie en is in sommige gevallen ongedaan gemaakt door stopzetting van de behandeling met TRICOR 200 mg. Patiënten met DILI hebben tekenen en symptomen ervaren, waaronder donkere urine, abnormale ontlasting, geelzucht, malaise, buikpijn, spierpijn, gewichtsverlies, pruritus en misselijkheid. Veel patiënten hadden gelijktijdige verhogingen van totaal bilirubine, serum-alaninetransaminase (ALAT) en aspartaattransaminase (AST). DILI is gekarakteriseerd als hepatocellulaire, chronisch actieve en cholestatische hepatitis, en cirrose is opgetreden in verband met chronische actieve hepatitis.
In klinische onderzoeken is fenofibraat in doses gelijk aan 96 mg tot 145 mg TRICOR 160 mg per dag in verband gebracht met verhogingen van de serum-ASAT of ALT. De incidentie van verhogingen van transaminasen kan dosisgerelateerd zijn [zie: ONGEWENSTE REACTIES ].
TRICOR 160 mg is gecontra-indiceerd bij patiënten met een actieve leverziekte, inclusief patiënten met primaire biliaire cirrose en onverklaarbare aanhoudende afwijkingen van de leverfunctie [zie CONTRA-INDICATIES ]. Controleer de leverfunctie van de patiënt, inclusief serum ALT, AST en totaal bilirubine, bij aanvang en periodiek voor de duur van de behandeling met TRICOR. Stop met TRICOR 200 mg als zich tekenen of symptomen van leverbeschadiging ontwikkelen of als verhoogde enzymspiegels aanhouden (ALAT of ASAT > 3 keer de bovengrens van normaal, of als het gepaard gaat met verhoging van bilirubine). Start TRICOR 160 mg niet opnieuw bij deze patiënten als er geen alternatieve verklaring is voor de leverbeschadiging.
Myopathie en rabdomyolyse
Fibraten verhogen het risico op myopathie en zijn in verband gebracht met rabdomyolyse. Het risico op ernstige spiertoxiciteit lijkt verhoogd te zijn bij oudere patiënten en bij patiënten met diabetes, nierinsufficiëntie of hypothyreoïdie.
Myopathie moet worden overwogen bij elke patiënt met diffuse myalgie, spiergevoeligheid of spierzwakte en/of duidelijke verhogingen van de creatinefosfokinase (CPK)-spiegels.
Patiënten moeten worden geadviseerd onverklaarbare spierpijn, gevoeligheid of zwakte onmiddellijk te melden, vooral als deze gepaard gaat met malaise of koorts. De CPK-spiegels moeten worden beoordeeld bij patiënten die deze symptomen melden, en de behandeling met TRICOR 200 mg moet worden stopgezet als er duidelijk verhoogde CPK-spiegels optreden of als myopathie/myositis wordt vermoed of gediagnosticeerd.
Gegevens uit observationele onderzoeken geven aan dat het risico op rabdomyolyse groter is wanneer fibraten, in het bijzonder gemfibrozil, gelijktijdig worden toegediend met een statine. De combinatie moet worden vermeden, tenzij het voordeel van verdere veranderingen in lipideniveaus waarschijnlijk opweegt tegen het verhoogde risico van deze combinatie van geneesmiddelen [zie KLINISCHE FARMACOLOGIE ].
Gevallen van myopathie, waaronder rabdomyolyse, zijn gemeld met fenofibraat gelijktijdig toegediend met colchicine, en voorzichtigheid is geboden bij het voorschrijven van fenofibraat met colchicine [zie DRUG-INTERACTIES ].
Serum creatinine
Verhogingen van het serumcreatinine zijn gemeld bij patiënten die fenofibraat gebruiken. Deze verhogingen keren meestal terug naar de uitgangswaarde na stopzetting van fenofibraat. De klinische betekenis van deze waarnemingen is niet bekend. Controleer de nierfunctie bij patiënten met een nierfunctiestoornis die TRICOR gebruiken. Controle van de nieren moet ook worden overwogen bij patiënten die TRICOR gebruiken met een risico op nierinsufficiëntie, zoals ouderen en patiënten met diabetes.
cholelithiasis
Fenofibraat kan, net als clofibraat en gemfibrozil, de cholesteroluitscheiding in de gal verhogen, wat kan leiden tot cholelithiasis. Als cholelithiasis wordt vermoed, zijn galblaasonderzoeken geïndiceerd. De behandeling met TRICOR 160 mg moet worden stopgezet als galstenen worden gevonden.

Coumarine anticoagulantia
Voorzichtigheid is geboden wanneer cumarine-anticoagulantia worden gegeven in combinatie met TRICOR 160 mg vanwege de versterking van de anticoagulerende effecten van het cumarine-type bij het verlengen van de protrombinetijd/internationale genormaliseerde ratio (PT/INR). Om bloedingscomplicaties te voorkomen, worden frequente controle van de PT/INR en dosisaanpassing van het antistollingsmiddel aanbevolen totdat de PT/INR gestabiliseerd is [zie DRUG-INTERACTIES ].
Pancreatitis
Pancreatitis is gemeld bij patiënten die fenofibraat, gemfibrozil en clofibraat gebruikten. Dit optreden kan duiden op een falen van de werkzaamheid bij patiënten met ernstige hypertriglyceridemie, een direct geneesmiddeleffect of een secundair fenomeen dat wordt gemedieerd door galwegensteen of slibvorming met obstructie van het gemeenschappelijke galkanaal.
Hematologische veranderingen
Milde tot matige dalingen van hemoglobine, hematocriet en witte bloedcellen zijn waargenomen bij patiënten na het starten van de behandeling met fenofibraat. Deze niveaus stabiliseren echter tijdens langdurige toediening. Trombocytopenie en agranulocytose zijn gemeld bij personen die met fenofibraat werden behandeld. Periodieke controle van het aantal rode en witte bloedcellen wordt aanbevolen tijdens de eerste 12 maanden van toediening van TRICOR.
Overgevoeligheidsreacties
Acute overgevoeligheid
Anafylaxie en angio-oedeem zijn postmarketing gemeld met fenofibraat. In sommige gevallen waren de reacties levensbedreigend en was een spoedbehandeling vereist. Als een patiënt tekenen of symptomen van een acute overgevoeligheidsreactie ontwikkelt, adviseer hem dan om onmiddellijk medische hulp in te roepen en de behandeling met fenofibraat te staken.
Vertraagde overgevoeligheid
Ernstige cutane bijwerkingen (SCAR), waaronder Stevens-Johnson-syndroom, toxische epidermale necrolyse en geneesmiddelreactie met eosinofilie en systemische symptomen (DRESS), zijn gemeld na het in de handel brengen, dagen tot weken na het starten met fenofibraat. De gevallen van DRESS gingen gepaard met huidreacties (zoals huiduitslag of exfoliatieve dermatitis) en een combinatie van eosinofilie, koorts, systemische orgaanaantasting (nier-, lever- of ademhalingsorganen). Stop met fenofibraat en behandel patiënten op de juiste manier als SCAR wordt vermoed.
Venotrombo-embolische ziekte
In de FIELD-studie werden longembolie (PE) en diepe veneuze trombose (DVT) vaker waargenomen in de met fenofibraat behandelde groep dan in de met placebo behandelde groep. Van de 9.795 patiënten die deelnamen aan FIELD, waren er 4.900 in de placebogroep en 4.895 in de fenofibraatgroep. Voor DVT waren er 48 voorvallen (1%) in de placebogroep en 67 (1%) in de fenofibraatgroep (p = 0,074); en voor PE waren er 32 (0,7%) voorvallen in de placebogroep en 53 (1%) in de fenofibraatgroep (p = 0,022).
In het Coronary Drug Project had een groter deel van de clofibraatgroep een definitieve of vermoedelijke fatale of niet-fatale longembolie of tromboflebitis dan de placebogroep (5,2% vs. 3,3% na vijf jaar; p
Paradoxale dalingen in HDL-cholesterolniveaus
Er zijn postmarketingmeldingen en klinische onderzoeken geweest van ernstige verlagingen van het HDL-cholesterolgehalte (tot 2 mg/dl) die optreden bij diabetische en niet-diabetici die begonnen met fibraattherapie. De afname van HDL-C wordt weerspiegeld door een afname van apolipoproteïne A1. Van deze afname is gemeld dat deze optreedt binnen 2 weken tot jaren na het starten van de behandeling met fibraat. De HDL-C-spiegels blijven laag totdat de fibraattherapie is stopgezet; de respons op het staken van de behandeling met fibraat is snel en aanhoudend. De klinische betekenis van deze afname van HDL-C is niet bekend. Het wordt aanbevolen om de HDL-C-spiegels binnen de eerste paar maanden na het starten van de fibraattherapie te controleren. Als een ernstig verlaagde HDL-C-spiegel wordt gedetecteerd, moet de behandeling met fibraat worden stopgezet en moet de HDL-C-spiegel worden gecontroleerd totdat deze weer op de uitgangswaarde is bereikt, en mag de behandeling met fibraat niet opnieuw worden gestart.
Niet-klinische toxicologie
Carcinogenese en mutagenese en aantasting van de vruchtbaarheid
Er zijn twee carcinogeniteitsstudies via de voeding uitgevoerd bij ratten met fenofibraat. In het eerste onderzoek van 24 maanden kregen Wistar-ratten een dosis fenofibraat van 10, 45 en 200 mg/kg/dag, ongeveer 0,3, 1 en 6 keer de maximaal aanbevolen dosis voor mensen (MRHD) van 300 mg fenofibraat per dag, wat overeenkomt met 145 mg TRICOR 160 mg per dag, gebaseerd op vergelijkingen van het lichaamsoppervlak. Bij een dosis van 200 mg/kg/dag (bij 6 maal de MRHD) was de incidentie van levercarcinomen significant verhoogd bij beide geslachten. Een statistisch significante toename van pancreascarcinomen werd waargenomen bij mannen bij 1 en 6 keer de MRHD; een toename van pancreasadenomen en goedaardige testiculaire interstitiële celtumoren werd waargenomen bij 6 keer de MRHD bij mannen. In een tweede carcinogeniteitsonderzoek van 24 maanden bij een andere rattenstam (Sprague-Dawley) veroorzaakten doses van 10 en 60 mg/kg/dag (0,3 en 2 maal de MRHD) een significante toename van de incidentie van acinaire adenomen van de alvleesklier bij beide geslachten en toename van testiculaire interstitiële celtumoren bij mannen bij 2 keer de MRHD.
Er werd een carcinogeniteitsonderzoek van 117 weken uitgevoerd bij ratten waarin drie geneesmiddelen werden vergeleken: fenofibraat 10 en 60 mg/kg/dag (0,3 en 2 keer de MRHD, gebaseerd op vergelijkingen van het lichaamsoppervlak), clofibraat (400 mg/kg/dag; de humane dosis) en gemfibrozil (250 mg/kg/dag; 2 maal de humane dosis, gebaseerd op mg/m² oppervlakte). Fenofibraat verhoogde acinaire adenomen van de pancreas bij beide geslachten. Clofibraat verhoogde hepatocellulair carcinoom en acinaire adenomen van de pancreas bij mannen en hepatische neoplastische knobbeltjes bij vrouwen. Gemfibrozil verhoogde hepatische neoplastische knobbeltjes bij mannen en vrouwen, terwijl alle drie de geneesmiddelen de testiculaire interstitiële celtumoren bij mannen deden toenemen.
In een onderzoek van 21 maanden bij CF-1-muizen verhoogde fenofibraat 10, 45 en 200 mg/kg/dag (ongeveer 0,2, 1 en 3 keer de MRHD, gebaseerd op vergelijkingen van het lichaamsoppervlak) de levercarcinomen significant in beide geslachten op 3 keer de MRHD. In een tweede onderzoek van 18 maanden met 10, 60 en 200 mg/kg/dag verhoogde fenofibraat de levercarcinomen bij mannelijke muizen en leveradenomen bij vrouwelijke muizen significant bij 3 keer de MRHD.
Elektronenmicroscopie-onderzoeken hebben peroxisomale proliferatie aangetoond na toediening van fenofibraat aan de rat. Er is geen adequaat onderzoek gedaan om peroxisoomproliferatie bij mensen te testen, maar veranderingen in peroxisoommorfologie en -aantallen zijn waargenomen bij mensen na behandeling met andere leden van de fibraatklasse wanneer leverbiopten werden vergeleken voor en na behandeling bij dezelfde persoon.
In de volgende tests is aangetoond dat fenofibraat geen mutageen potentieel heeft: Ames, muislymfoom, chromosomale aberratie en ongeplande DNA-synthese in primaire hepatocyten van ratten.
In vruchtbaarheidsonderzoeken kregen ratten orale dieetdoses fenofibraat, mannetjes kregen 61 dagen voorafgaand aan het paren en vrouwtjes 15 dagen voorafgaand aan het paren tot aan het spenen, wat resulteerde in geen nadelig effect op de vruchtbaarheid bij doses tot 300 mg/kg/dag (10 maal de MRHD, gebaseerd op vergelijkingen van het lichaamsoppervlak).
Gebruik bij specifieke populaties
Zwangerschap
Risico Samenvatting
Beperkte beschikbare gegevens over het gebruik van fenofibraat bij zwangere vrouwen zijn onvoldoende om een geneesmiddelgerelateerd risico op ernstige geboorteafwijkingen, miskraam of ongunstige maternale of foetale uitkomsten vast te stellen. In reproductiestudies bij dieren werd geen bewijs van embryo-foetale toxiciteit waargenomen bij orale toediening van fenofibraat bij ratten en konijnen tijdens de organogenese in doses lager dan of gelijk aan de maximaal aanbevolen klinische dosis van 145 mg per dag, gebaseerd op het lichaamsoppervlak (mg/ m²). Nadelige reproductieresultaten traden op bij hogere doses in aanwezigheid van maternale toxiciteit (zie: Gegevens ). TRICOR 200 mg mag alleen tijdens de zwangerschap worden gebruikt als het mogelijke voordeel opweegt tegen het mogelijke risico voor de foetus.
Het geschatte achtergrondrisico van ernstige geboorteafwijkingen en miskraam voor de aangegeven populatie is niet bekend. In de algemene bevolking van de VS is het geschatte achtergrondrisico van ernstige geboorteafwijkingen en miskraam bij klinisch erkende zwangerschappen respectievelijk 2-4% en 15-20%.
Gegevens
Dierlijke gegevens
Bij drachtige ratten die orale voedingsdoses van 14, 127 en 361 mg/kg/dag kregen vanaf dag 6-15 van de dracht tijdens de periode van organogenese, werden geen nadelige ontwikkelingsbevindingen waargenomen bij 14 mg/kg/dag (minder dan de klinische blootstelling bij de maximaal aanbevolen dosis voor mensen [MRHD] van 300 mg fenofibraat per dag, overeenkomend met 145 mg TRICOR per dag, op basis van vergelijkingen van het lichaamsoppervlak). Verhoogde foetale skeletmisvormingen werden waargenomen bij maternale toxische doses (361 mg/kg/dag, overeenkomend met 12 keer de klinische blootstelling bij de MRHD) die de gewichtstoename van de moeder significant onderdrukte.
Bij drachtige konijnen die orale toedieningsdoses van 15, 150 en 300 mg/kg/dag kregen vanaf dag 618 van de dracht tijdens de periode van organogenese en die lieten bevallen, werden geen nadelige ontwikkelingsbevindingen waargenomen bij 15 mg/kg/dag (een dosis die benadert de klinische blootstelling bij de MRHD, gebaseerd op vergelijkingen van het lichaamsoppervlak). Geaborteerde nesten werden waargenomen bij maternale toxische doses (≥ 150 mg/kg/dag, overeenkomend met ≥ 10 keer de klinische blootstelling bij de MRHD) die de toename van het lichaamsgewicht van de moeder onderdrukten.
Bij drachtige ratten die orale dieetdoses van 15, 75 en 300 mg/kg/dag kregen vanaf dag 15 van de dracht tot dag 21 van de lactatie (spenen), werden geen nadelige ontwikkelingseffecten waargenomen bij 15 mg/kg/dag (minder dan de klinische blootstelling bij de MRHD, op basis van vergelijkingen van het lichaamsoppervlak), ondanks maternale toxiciteit (verminderde gewichtstoename). Post-implantatieverlies werd waargenomen bij ≥ 75 mg/kg/dag (≥ 2 maal de klinische blootstelling bij de MRHD) in aanwezigheid van maternale toxiciteit (verminderde gewichtstoename). Een verminderde overleving van de jongen werd waargenomen bij 300 mg/kg/dag (10 maal de klinische blootstelling bij de MRHD), wat in verband werd gebracht met een verminderde gewichtstoename van de moeder/verwaarlozing door de moeder.
Borstvoeding
Risico Samenvatting
Er is geen informatie beschikbaar over de aanwezigheid van fenofibraat in moedermelk, de effecten van het geneesmiddel op de zuigeling die borstvoeding krijgt of de effecten op de melkproductie. Fenofibraat is aanwezig in de melk van ratten en is daarom waarschijnlijk aanwezig in de moedermelk. Vanwege de mogelijkheid van ernstige bijwerkingen bij zuigelingen die borstvoeding krijgen, zoals verstoring van het lipidenmetabolisme bij zuigelingen, mogen vrouwen geen borstvoeding geven tijdens de behandeling met TRICOR 200 mg en gedurende 5 dagen na de laatste dosis [zie CONTRA-INDICATIES ].
Pediatrisch gebruik
De veiligheid en werkzaamheid zijn niet vastgesteld bij pediatrische patiënten.
Geriatrisch gebruik
Van fenofibrinezuur is bekend dat het grotendeels door de nieren wordt uitgescheiden en het risico op bijwerkingen van dit geneesmiddel kan groter zijn bij patiënten met een verminderde nierfunctie. Blootstelling aan fenofibrinezuur wordt niet beïnvloed door leeftijd. Aangezien oudere patiënten een hogere incidentie van nierinsufficiëntie hebben, moet de dosiskeuze voor ouderen worden gemaakt op basis van de nierfunctie [zie: DOSERING EN ADMINISTRATIE en KLINISCHE FARMACOLOGIE ]. Bij oudere patiënten met een normale nierfunctie is geen dosisaanpassing nodig. Overweeg de nierfunctie te controleren bij oudere patiënten die TRICOR gebruiken.
Nierfunctiestoornis
Het gebruik van TRICOR 160 mg moet worden vermeden bij patiënten met een ernstige nierfunctiestoornis [zie: CONTRA-INDICATIES ]. Dosisverlaging is vereist bij patiënten met lichte tot matige nierinsufficiëntie [zie: DOSERING EN ADMINISTRATIE en KLINISCHE FARMACOLOGIE ]. Controle van de nierfunctie bij patiënten met een nierfunctiestoornis wordt aanbevolen.
Leverfunctiestoornis
Het gebruik van TRICOR 200 mg is niet geëvalueerd bij personen met een leverfunctiestoornis [zie: CONTRA-INDICATIES en KLINISCHE FARMACOLOGIE ].
OVERDOSERING
Er is geen specifieke behandeling voor overdosering met TRICOR. Algemene ondersteunende zorg voor de patiënt is geïndiceerd, inclusief monitoring van vitale functies en observatie van de klinische status, mocht zich een overdosis voordoen. Indien geïndiceerd, moet de eliminatie van niet-geabsorbeerd geneesmiddel worden bereikt door braken of maagspoeling; de gebruikelijke voorzorgsmaatregelen moeten in acht worden genomen om de luchtweg te behouden. Omdat fenofibrinezuur sterk gebonden is aan plasma-eiwitten, dient hemodialyse niet te worden overwogen.
CONTRA-INDICATIES
TRICOR 200 mg is gecontra-indiceerd bij:
- patiënten met ernstige nierinsufficiëntie, inclusief diegenen die dialyse ondergaan [zie: KLINISCHE FARMACOLOGIE ].
- patiënten met een actieve leverziekte, inclusief die met primaire biliaire cirrose en onverklaarbare aanhoudende leverfunctieafwijkingen [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
- patiënten met een reeds bestaande galblaasaandoening [zie WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
- zogende moeders [zie Gebruik bij specifieke populaties ].
- patiënten met bekende overgevoeligheid voor fenofibraat of fenofibrinezuur [zie: WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ].
KLINISCHE FARMACOLOGIE
Werkingsmechanisme
Het actieve deel van TRICOR is fenofibrinezuur. De farmacologische effecten van fenofibrinezuur bij zowel dieren als mensen zijn uitgebreid onderzocht door middel van orale toediening van fenofibraat.
De lipidenmodificerende effecten van fenofibrinezuur die in de klinische praktijk worden waargenomen, zijn in vivo verklaard bij transgene muizen en in vitro in humane hepatocytculturen door de activering van peroxisoomproliferator-geactiveerde receptor α (PPARα). Door dit mechanisme verhoogt fenofibraat de lipolyse en eliminatie van triglyceriderijke deeltjes uit het plasma door lipoproteïnelipase te activeren en de productie van apoproteïne C-III (een remmer van lipoproteïnelipase-activiteit) te verminderen.
De resulterende afname van TG veroorzaakt een verandering in de grootte en samenstelling van LDL van kleine, dichte deeltjes (waarvan wordt aangenomen dat ze atherogeen zijn vanwege hun gevoeligheid voor oxidatie), tot grote drijvende deeltjes. Deze grotere deeltjes hebben een grotere affiniteit voor cholesterolreceptoren en worden snel afgebroken. Activering van PPARα induceert ook een toename van de synthese van apolipoproteïnen AI, A-II en HDL-cholesterol.
Fenofibraat verlaagt ook de serumurinezuurspiegels bij hyperurikemische en normale personen door de urinaire excretie van urinezuur te verhogen.
farmacodynamiek
Verschillende klinische onderzoeken hebben aangetoond dat verhoogde niveaus van totaal-C, LDL-C en apo B, een LDL-membraancomplex, geassocieerd zijn met humane atherosclerose. Evenzo worden verlaagde niveaus van HDL-C en zijn transportcomplex, apolipoproteïne A (apo AI en apo AII) geassocieerd met de ontwikkeling van atherosclerose. Epidemiologisch onderzoek heeft aangetoond dat cardiovasculaire morbiditeit en mortaliteit direct variëren met het niveau van totaal-C, LDL-C en TG, en omgekeerd met het niveau van HDL-C. Het onafhankelijke effect van het verhogen van HDL-C of het verlagen van triglyceriden (TG) op het risico op cardiovasculaire morbiditeit en mortaliteit is niet vastgesteld.
Fenofibrinezuur, de actieve metaboliet van fenofibraat, veroorzaakt bij behandelde patiënten verlagingen van totaal cholesterol, LDL-cholesterol, apolipoproteïne B, totale triglyceriden en triglyceridenrijk lipoproteïne (VLDL). Bovendien resulteert behandeling met fenofibraat in verhogingen van high-density lipoproteïne (HDL) en apolipoproteïnen apoAI en apoAII.
Farmacokinetiek
Plasmaconcentraties van fenofibrinezuur na toediening van drie 48 mg of één 145 mg tabletten zijn onder gevoede omstandigheden gelijk aan één 200 mg gemicroniseerde fenofibraatcapsule.
Fenofibraat is een pro-drug van de actieve chemische groep fenofibrinezuur. Fenofibraat wordt door esterhydrolyse in het lichaam omgezet in fenofibrinezuur, het actieve bestanddeel dat in de bloedsomloop kan worden gemeten.
Absorptie
De absolute biologische beschikbaarheid van fenofibraat kan niet worden bepaald, aangezien de verbinding vrijwel onoplosbaar is in waterige media die geschikt zijn voor injectie. Fenofibraat wordt echter goed geabsorbeerd uit het maagdarmkanaal. Na orale toediening aan gezonde vrijwilligers verscheen ongeveer 60% van een enkele dosis radioactief gelabeld fenofibraat in de urine, voornamelijk als fenofibrinezuur en zijn glucuronaatconjugaat, en 25% werd uitgescheiden in de feces. Piekplasmaspiegels van fenofibrinezuur treden op binnen 6 tot 8 uur na toediening.
Blootstelling aan fenofibrinezuur in plasma, zoals gemeten door Cmax en AUC, is niet significant verschillend wanneer een enkelvoudige dosis fenofibraat van 145 mg wordt toegediend onder nuchtere of niet-nuchtere omstandigheden.
Verdeling
Na meervoudige dosering van fenofibraat wordt de steady state van fenofibrinezuur binnen 9 dagen bereikt. Plasmaconcentraties van fenofibrinezuur bij steady state zijn ongeveer het dubbele van die na een enkele dosis. De serumeiwitbinding was ongeveer 99% bij normale en hyperlipidemische proefpersonen.
Metabolisme
Na orale toediening wordt fenofibraat snel gehydrolyseerd door esterasen tot de actieve metaboliet fenofibrinezuur; er wordt geen onveranderd fenofibraat gedetecteerd in plasma.
Fenofibrinezuur wordt voornamelijk geconjugeerd met glucuronzuur en vervolgens uitgescheiden in de urine. Een kleine hoeveelheid fenofibrinezuur wordt aan de carbonylgroep gereduceerd tot een benzhydrolmetaboliet die op zijn beurt wordt geconjugeerd met glucuronzuur en wordt uitgescheiden in de urine.
In vivo metabolismegegevens geven aan dat fenofibraat noch fenofibrinezuur in significante mate oxidatief metabolisme ondergaan (bijv. cytochroom P450).
Eliminatie
Na absorptie wordt fenofibraat voornamelijk in de urine uitgescheiden in de vorm van metabolieten, voornamelijk fenofibrinezuur en fenofibrinezuurglucuronide. Na toediening van radioactief gelabeld fenofibraat verscheen ongeveer 60% van de dosis in de urine en 25% werd uitgescheiden in de feces.
Fenofibrinezuur wordt geëlimineerd met een halfwaardetijd van 20 uur, waardoor een eenmaal daagse dosering mogelijk is.
Speciale populaties
Geriatrie
Bij oudere vrijwilligers in de leeftijd van 77 tot 87 jaar was de orale klaring van fenofibrinezuur na een enkelvoudige orale dosis fenofibraat 1,2 l/u, vergeleken met 1,1 l/u bij jonge volwassenen. Dit geeft aan dat een vergelijkbaar doseringsschema kan worden gebruikt bij ouderen met een normale nierfunctie, zonder toenemende accumulatie van het geneesmiddel of metabolieten [zie DOSERING EN ADMINISTRATIE en Gebruik bij specifieke populaties ].
Kindergeneeskunde
De farmacokinetiek van TRICOR 160 mg is niet onderzocht bij pediatrische populaties.
Geslacht
Er is geen farmacokinetisch verschil tussen mannen en vrouwen waargenomen voor fenofibraat.
Ras
De invloed van ras op de farmacokinetiek van fenofibraat is niet onderzocht, maar fenofibraat wordt niet gemetaboliseerd door enzymen waarvan bekend is dat ze interetnische variabiliteit vertonen.
Nierfunctiestoornis
De farmacokinetiek van fenofibrinezuur werd onderzocht bij patiënten met lichte, matige en ernstige nierinsufficiëntie. Patiënten met een ernstige nierfunctiestoornis (geschatte glomerulaire filtratiesnelheid [eGFR] DOSERING EN ADMINISTRATIE ].
Leverfunctiestoornis
Er zijn geen farmacokinetische onderzoeken uitgevoerd bij patiënten met leverinsufficiëntie.
Geneesmiddel-geneesmiddelinteracties
In vitro-onderzoeken met humane levermicrosomen geven aan dat fenofibraat en fenofibrinezuur geen remmers zijn van de cytochroom (CYP) P450-isovormen CYP3A4, CYP2D6, CYP2E1 of CYP1A2. Het zijn zwakke remmers van CYP2C8, CYP2C19 en CYP2A6, en milde tot matige remmers van CYP2C9 bij therapeutische concentraties.
Tabel 2 beschrijft de effecten van gelijktijdig toegediende geneesmiddelen op de systemische blootstelling aan fenofibrinezuur. Tabel 3 beschrijft de effecten van gelijktijdig toegediend fenofibraat of fenofibrinezuur op andere geneesmiddelen.
Klinische studies
Primaire hypercholesterolemie (heterozygote familiale en niet-familiale) en gemengde dyslipidemie
De effecten van fenofibraat in een dosis equivalent aan 145 mg TRICOR (fenofibraattabletten) per dag werden beoordeeld op basis van vier gerandomiseerde, placebogecontroleerde, dubbelblinde onderzoeken met parallelle groepen met patiënten met de volgende gemiddelde lipidewaarden bij baseline: totaal-C 306,9 mg/dL; LDL-C 213,8 mg/dL; HDL-C 52,3 mg/dL; en triglyceriden 191,0 mg/dL. TRICOR 200 mg-therapie verlaagde de LDL-C, Total-C en de LDL-C/HDL-C-ratio. TRICOR-therapie verlaagde ook de triglyceriden en verhoogde HDL-C (zie tabel 4).
Bij een subset van de proefpersonen werden metingen van apo B uitgevoerd. Behandeling met TRICOR 200 mg verminderde apo B significant van baseline tot eindpunt in vergelijking met placebo (respectievelijk -25,1% vs. 2,4%, p
Ernstige hypertriglyceridemie
De effecten van fenofibraat op serumtriglyceriden werden onderzocht in twee gerandomiseerde, dubbelblinde, placebogecontroleerde klinische onderzoeken bij 147 hypertriglyceridemische patiënten. Patiënten werden gedurende acht weken behandeld volgens protocollen die alleen verschilden doordat één patiënt werd opgenomen met baseline TG-waarden van 500 tot 1500 mg/dL en de andere TG-waarden van 350 tot 500 mg/dL. Bij patiënten met hypertriglyceridemie en normale cholesterolemie met of zonder hyperchylomicronemie, verminderde behandeling met fenofibraat in doseringen equivalent aan TRICOR 145 mg per dag voornamelijk zeer lage dichtheid lipoproteïne (VLDL) triglyceriden en VLDL-cholesterol. Behandeling van patiënten met verhoogde triglyceriden resulteert vaak in een verhoging van LDL-C (zie tabel 5).
Het effect van TRICOR 160 mg op cardiovasculaire morbiditeit en mortaliteit is niet vastgesteld.
PATIËNT INFORMATIE
Patiënten moeten worden geadviseerd:
- van de mogelijke voordelen en risico's van TRICOR.
- TRICOR niet gebruiken als er een bekende overgevoeligheid is voor fenofibraat of fenofibrinezuur.
- van medicijnen die niet in combinatie met TRICOR mogen worden ingenomen.
- dat als ze cumarine-anticoagulantia gebruiken, TRICOR 160 mg hun anticoagulerende effect kan versterken en dat verhoogde controle nodig kan zijn.
- om een geschikt lipidenmodificerend dieet te blijven volgen terwijl u TRICOR gebruikt.
- om TRICOR 160 mg eenmaal daags in te nemen, ongeacht voedsel, in de voorgeschreven dosis, waarbij elke tablet in zijn geheel wordt doorgeslikt.
- om terug te keren naar het kantoor van hun arts voor routinematige controle.
- om hun arts te informeren over alle medicijnen, supplementen en kruidenpreparaten die ze nemen en elke verandering in hun medische toestand. Patiënten moeten ook worden geadviseerd om hun arts die een nieuw medicijn voorschrijft, te informeren dat ze TRICOR gebruiken.
- om hun arts te informeren over symptomen van leverbeschadiging (bijv. geelzucht, buikpijn, misselijkheid, malaise, donkere urine, abnormale ontlasting, pruritus); elke spierpijn, gevoeligheid of zwakte; of andere nieuwe symptomen.
- geen borstvoeding geven tijdens de behandeling met TRICOR en gedurende 5 dagen na de laatste dosis.